Hiver 2025 (Volume 35, numéro 4)
De la polyarthrite à la nodulose agressive : une évolution inhabituelle
de la PR
Par Kavya Mulgund et Tripti Papneja, M.D., FRCPC
Télécharger la version PDF
|
Présentation de cas
Une femme de 61 ans souffrant d'hypertension s'est présentée pour la première fois en 2011, se plaignant de douleurs articulaires, de raideurs matinales et de fatigue depuis plusieurs semaines. L'examen a révélé un gonflement de plusieurs articulations métacarpophalangiennes (MCP) sans signes extra-articulaires. Les analyses de laboratoire ont révélé un taux d'hémoglobine de 88 g/L, un taux de ferritine de 8 µg/L, une vitesse de sédimentation de 71 mm/h, un taux de protéine C-réactive de 34,8 mg/L, un facteur rhumatoïde de 299 UI/ml et un taux d'ANA de 1:640. L'anti-CCP était négatif. Elle a reçu un diagnostic de polyarthrite rhumatoïde (PR) séropositive et a commencé un traitement par méthotrexate et hydroxychloroquine, auquel elle a bien répondu. Sa PR est entrée en rémission en 2013 et elle a choisi d'arrêter le traitement par des médicaments antirhumatismaux modificateurs de la maladie (ARMM). Entre 2013 et 2024, elle a été traitée pour une ostéoporose, passant d'un traitement aux bisphosphonates à des injections de dénosumab. Elle a continué à prendre du valsartan et de l'amlodipine pour son hypertension. Elle a développé le phénomène de Raynaud en février 2023.
Elle était au Pakistan d'avril à mai 2024 et a commencé à se sentir très mal, avec de la fièvre, une fatigue marquée et des douleurs/gonflements articulaires. Elle ne pouvait plus bouger son coude droit. Elle a commencé un traitement à base de prednisone à raison de 60 mg par jour, à réduire de 5 mg tous les deux jours. En juin 2024, elle a développé des nodules sur les mains (Figure 1) et a signalé une anorexie avec une perte de poids de 9 kg, une fièvre intermittente de faible intensité, une toux et une xérostomie. L'examen a révélé 6 articulations enflées, touchant principalement les articulations MCP et interphalangiennes proximales (IPP), ainsi que de multiples nodules discrets et sensibles sur la face dorsale et palmaire des deux mains.
Les analyses sanguines ont montré les résultats suivants : hémoglobine 114 g/L, leucocytes 4,1 ×109/L, plaquettes 219 ×109/L, vitesse de sédimentation de 40 mm/h, protéine C-réactive de 1,3 mg/L, ANA persistants ≥ 1:640 mouchetés, SSA/SSB positifs (> 8,0, la normale étant < 1,0), RNP légèrement positifs (2,0, la normale étant < 1,0), faibles taux de complément (C3 0,80 g/L, C4 0,14 g/L) et tests anti-ADN double brin, hépatite B/C, VIH et Quantiferon Gold TB négatifs. Un diagnostic provisoire de poussée de PR a été posé et un traitement à base de prednisone et de léflunomide a été instauré.
|
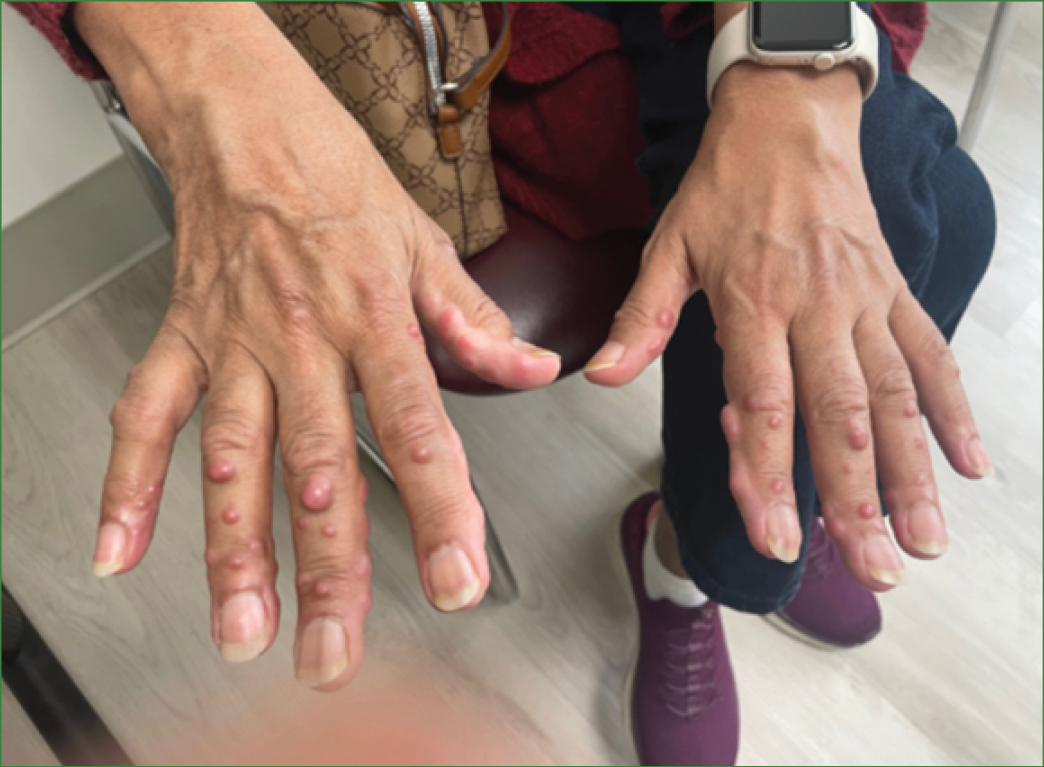
Figure 1. Lésions cutanées papulonodulaires sur les mains de notre patiente.
La biopsie à l'emporte-pièce d'un nodule de la main (août 2024) a révélé une prolifération dermique d'histiocytes et de cellules géantes multinucléées contenant un cytoplasme éosinophile « en verre dépoli » avec un stroma sclérotique, confirmant le diagnostic de réticulohistiocytose multicentrique (RHM). La biopsie d'une lésion sur son dos était compatible avec un granulome annulaire.
Compte tenu du diagnostic de RHM, un dépistage du cancer a été effectué. La coloscopie, la mammographie et la tomodensitométrie cérébrale étaient normales. La tomodensitométrie abdominale/pelvienne a révélé des kystes hépatiques et rénaux. La tomodensitométrie thoracique a montré une maladie pulmonaire interstitielle kystique avec de multiples kystes à paroi mince, correspondant le plus à une pneumonie interstitielle lymphoïde. Son test de fonction pulmonaire a révélé des anomalies restrictives. Elle a subi une échographie endobronchique le 6 novembre afin de prélever des échantillons des ganglions lymphatiques médiastinaux, dont les résultats sont en attente. L'ORL surveille une masse paratrachéale droite de 3,7 x 3,0 cm, vraisemblablement due à une hypertrophie de la thyroïde.
Discussion
La réticulohistiocytose multicentrique (RHM) est une histiocytose rare, multisystémique, de classe IIb, non liée aux cellules de Langerhans, caractérisée par une prolifération granulomateuse du système phagocytaire mononucléaire. Elle se manifeste par une polyarthrite destructrice et des lésions cutanées papulonodulaires. La polyarthrite est souvent le premier symptôme et le plus visible, évoluant vers une arthrite destructrice et un handicap. Nous présentons un cas de RHM et passons en revue les publications actuelles sur ses caractéristiques cliniques, son approche diagnostique et sa prise en charge.
Épidémiologie
La RHM est une maladie rare avec un peu plus de 300 cas signalés dans le monde. L'incidence et la prévalence réelles de cette maladie sont inconnues. Elle touche principalement les femmes caucasiennes âgées de 50 à 60 ans, avec un rapport femmes/hommes de 3:1.
La RHM est souvent associé à des maladies auto-immunes et à des tumeurs malignes internes. Une étude de la Mayo Clinic (1980-2017) portant sur 24 cas a rapporté des maladies autoimmunes dans 29 % des cas, des tumeurs malignes dans 25 % des cas et un taux de survie à cinq ans de 85 % (IC à 95 : 74-100 %)2. La RHM coexiste fréquemment avec des maladies autoimmunes, notamment le syndrome de Sjögren, le lupus érythémateux disséminé, la sclérose systémique, la dermatomyosite, la maladie cœliaque et la cirrhose biliaire primitive.
Bien que les cas de RHM aient été associés à presque tous les types de cancer, tant solides qu'hématologiques, les tumeurs malignes les plus couramment observées sont les carcinomes du poumon, de l'estomac, du sein, du col de l'utérus, du côlon et de l'ovaire. La question de savoir si la réticulohistiocytose multicentrique est un véritable trouble paranéoplasique est controversée, car aucun type de cancer cohérent n'a été associé à la RHM. De plus, comme la réticulohistiocytose multicentrique est très rare, l'association avec le cancer peut être fortuite. En outre, aucune corrélation entre l'ablation du cancer et la disparition ou l'amélioration de la réticulohistiocytose multicentrique n'a été établie.
Caractéristiques cliniques
Chez la moitié des patients, le premier signe de la maladie est l'arthrite. Chez un quart d'entre eux, ce sont des papules et des nodules qui apparaissent en premier. Les autres développent simultanément des manifestations cutanées et articulaires. Les manifestations articulaires les plus fréquentes comprennent une arthrite inflammatoire symétrique et érosive touchant principalement les mains, mais la RHM peut également toucher les coudes, les épaules, les hanches, les genoux et les pieds. Si elle n'est pas traitée, elle peut entraîner une arthropathie progressivement déformante et destructrice, notamment des contractures et une arthrite mutilante.
Les lésions cutanées apparaissent généralement dans les trois ans suivant l'apparition de l'arthrite, sous forme de papulonodules acraux jaunâtres à brun rougeâtre. Ces lésions surviennent le plus souvent sur la moitié supérieure du corps, en particulier le visage, les oreilles, les muqueuses (lèvres, langue, gencives, narines, gorge, paupières), les mains et les avant-bras. Leur taille varie de 1 à 2 mm à plusieurs centimètres de diamètre et elles apparaissent de manière isolée ou en grappes ou en groupes, avec un aspect pavé. Des lésions muqueuses sont présentes dans environ 50 % des cas. Les lésions cutanées peuvent entraîner la destruction du cartilage autour des oreilles et du nez. Les lésions sont généralement asymptomatiques, mais un tiers des patients se plaignent de prurit. Les lésions cutanées papulo-nodulaires périunguéales sont pathognomoniques et fusionnent souvent pour former l'aspect classique de « perles de corail » ou de « collier de perles ». Chez notre patient, les nodules sont apparus dix ans après les premiers symptômes articulaires.
Les symptômes systémiques courants comprennent la fièvre, un malaise général et une perte de poids, souvent accompagnés d'une vitesse de sédimentation élevée, d'une anémie et d'une hypercholestérolémie1. L'atteinte systémique peut inclure des épanchements pleuraux ou péricardiques, une insuffisance cardiaque, une lymphadénopathie mésentérique et des lésions urogénitales3.
Environ un tiers présentent des sérologies auto-immunes (anti-Ro, anti-CCP, ANA). L'histopathologie révèle des infiltrats lymphohistiocytaires avec des cellules géantes multinucléées contenant un cytoplasme éosinophile en verre dépoli2. La pathogenèse implique l'activation des monocytes/macrophages et l'activité ostéoclastique.
En cas de suspicion de RHM, les tests diagnostiques présentés dans le tableau 1 doivent être envisagés.

Approches thérapeutiques
Le traitement vise à contrôler l'inflammation et à prévenir la destruction articulaire. La RHM étant une maladie rare, il n'existe pas de directives thérapeutiques standardisées. Les traitements comprennent les corticostéroïdes, les MAMM tels que le méthotrexate, l'azathioprine, la cyclosporine, le cyclophosphamide, le chlorambucil, le tacrolimus topique et le rituximab4-6. Tariq et ses collaborateurs ont rapporté que le méthotrexate contrôlait l'arthrite chez 28 % des patients et les lésions cutanées chez 38 %, tandis que le cyclophosphamide permettait d'obtenir une rémission complète chez 20 % des patients et une amélioration partielle chez 40 à 45 % d'entre eux7. Les agents anti-TNF, l'anakinra, les bisphosphonates, les inhibiteurs de Janus Kinase (JAK) (upadacitinib) et le tocilizumab constituent des options supplémentaires8.
Chez de nombreux patients, la réticulocytose multicentrique peut entrer en rémission après une durée moyenne de 8 ans; cependant, à ce stade, une destruction articulaire considérable peut déjà s'être produite. Une arthrite mutilante peut se développer dans 50 % des cas. Les patients se retrouvent alors avec des articulations déformées et invalidantes et un visage défiguré. Un diagnostic précoce et un traitement rapide par ARMM sont essentiels, car la RHM suit souvent une évolution agressive et érosive qui conduit à la destruction articulaire si elle n'est pas traitée.
Retour au cas clinique
Malgré l'intensification du traitement avec l'ajout de prednisone, d'hydroxychloroquine, de léflunomide et de méthotrexate en septembre 2024, elle a continué à souffrir de fatigue persistante, d'arthrite et de nodules progressifs. En août 2025, un traitement par adalimumab a été instauré. Au moment de la rédaction du présent rapport, elle a suivi huit semaines de traitement par adalimumab sans amélioration significative des symptômes rhumatologiques ou dermatologiques. Un changement de traitement vers le tocilizumab est envisagé.
Conclusion
La RHM est une maladie systémique rare mais grave qui se caractérise par des manifestations cutanées et articulaires distinctives. Son association avec des tumeurs malignes et des maladies autoimmunes nécessite une évaluation approfondie. Un diagnostic précoce et la mise en place rapide d'un traitement immunosuppresseur ou biologique améliorent les résultats.
Kavya Mulgund
Étudiante, B. Sc. Santé,
Université Queen’s
Kingston (Ontario)
Tripti Papneja, M.D., FRCPC
Professeure clinicienne,
Université de Toronto
William Osler Hospital
Brampton (Ontario)
Glossaire :
ANA: anticorps antinucléaire
Anti-PCC: anticorps anti-peptide cituliné cyclique
Anti-dsDNA: anti-ADN double brin
Anti-Ro: anticorps anti-Ro, aussi appelés anticorps anti-SSA/Ro
PCR : protéine C-réactive
ORL : spécialiste des oreilles, du nez et de la gorge
VSE : vitesse de sédimentation
VIH : virus de l'immunodéficience humaine
RNP : ribonucléoprotéine
SSA/SSB: anticorps A du syndrôme de Sjögren's/anticoprs B du syndrôme de Sjögren
TB: tuberculose
GB : globule blancp>
Références :
1. Ashaolu O, Ng S, Smale S, Hughes J. Multicentric Reticulohistiocytosis–A rare and disabling disease. Clinical Case Reports. 2023; 11(9):e7846. https://doi.org/10.1002/ccr3.7846
2. Sanchez-Alvarez C, Singh Sandhu A, Crowson CS, et coll. Multicentric reticulohistiocytosis: the Mayo Clinic experience (1980–2017), Rheumatology. 2020; 59(8):1898-1905. https://doi.org/10.1093/rheumatology/kez555
3. Mariotti E, Corrà A, Lemmi E, et coll. Multicentric Reticulohistiocytosis Associated with an Early Form of Systemic Lupus Erythematosus: A Case Report of a Rare Disease, with Mini Review of the Literature. Journal of Clinical Medicine. 2022; 11(21):6529-6529. doi:https://doi.org/10.3390/jcm11216529
4. Bin FB. Multicentric reticulohistiocytosis in a Malaysian Chinese lady: A case report and review of literature. Dermatology Online Journal. 2009;15(1). doi:https://doi.org/10.5070/d34wm552ck
5. Liu YH, Fang K. Multicentric reticulohistiocytosis with generalized systemic involvement. Clinical and Experimental Dermatology. 2004; 29(4):373-376. doi:https://doi.org/10.1111/j.1365-2230.2004.01531.x
6. Lim K, D’Souza J, Vasquez JB, et coll. Looks Can Be Deceiving: A Case Report on Multicentric Reticulohistiocytosis Successfully Treated with Rituximab. Cureus. Published online May 3, 2017. doi:https://doi.org/10.7759/cureus.1220
7. Tariq S, Hugenberg ST, Hirano-Ali SA, et coll. Multicentric reticulohistiocytosis (MRH): case report with review of literature between 1991 and 2014 with in depth analysis of various treatment regimens and outcomes. SpringerPlus.2016; 5:180. doi:https://doi.org/10.1186/s40064-016-1874-5
8. Pacheco-Tena C, Reyes-Cordero G, Ochoa-Albíztegui R, et coll. Treatment of Multicentric Reticulohistiocytosis With Tocilizumab. Journal of Clinical Rheumatology. 2013; 19(5):272-276. doi:https://doi.org/10.1097/rhu.0b013e31829cf32b
|
-FR-v1.gif)


-FR-v1.gif)
