Printemps 2024 (Volume 34, numéro 1)
Un cas d’inflammation persistante
Par Jason An, M.D., et Dilan Dissanayake M.D., Ph. D.
Télécharger la version PDF
|
Présentation du cas
|
Un homme de 78 ans a été référé en rhumatologie pour une suspicion de polychondrite récidivante. Ses antécédents médicaux étaient marqués par un syndrome myélodysplasique (SMD) confirmé par une biopsie de la moelle osseuse en hématologie.
Il a remarqué pour la première fois une rougeur, une douleur et un gonflement du nez et de l’oreille droite quatre mois plus tôt. Il s’est rendu dans de nombreuses cliniques sans rendez-vous et n’a pas réagi aux nombreux traitements antibiotiques. Il a répondu à un court traitement de stéroïdes, mais les symptômes sont réapparus à la fin du traitement.
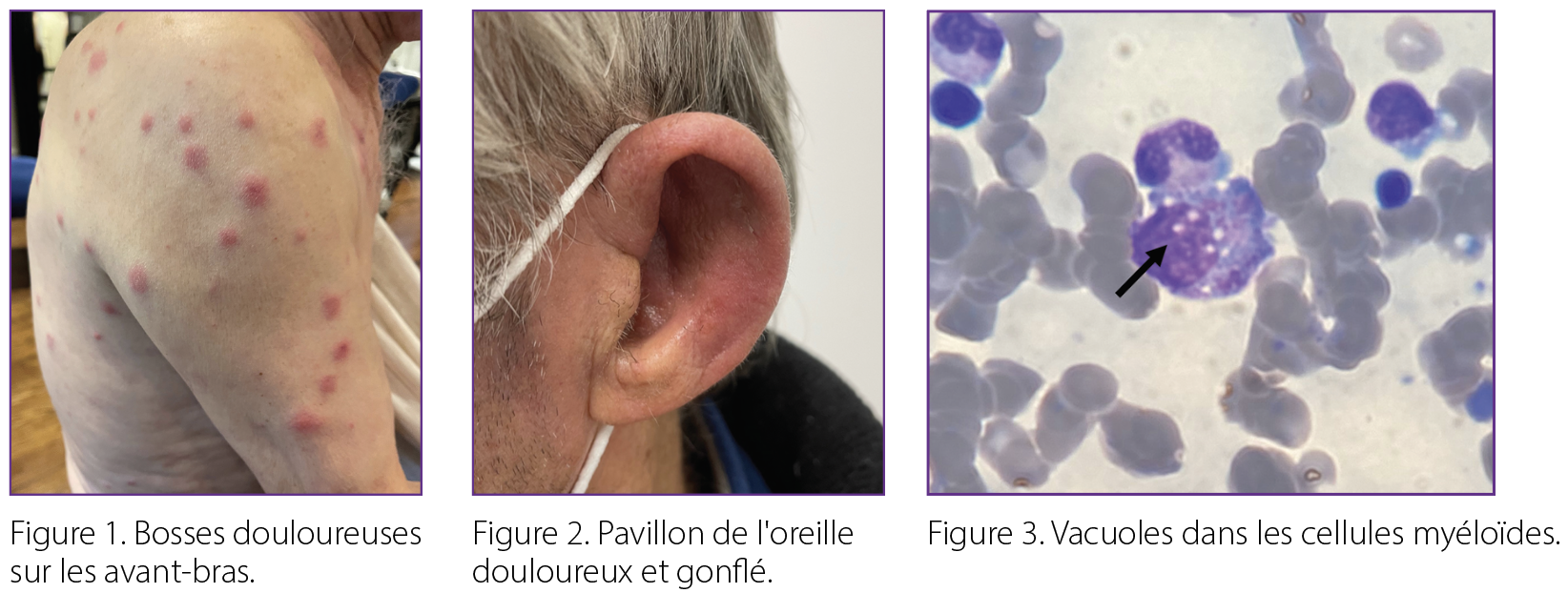
Il présentait également de nombreux autres symptômes inexpliqués au cours des mois précédents. Il s’agissait notamment de fièvres récurrentes allant jusqu’à 39 °C, d’une perte de poids involontaire de 15 livres, des yeux rouges et douloureux, d’une sécheresse buccale et de douleurs dans plusieurs articulations des mains. Il a remarqué des bosses douloureuses aux avant-bras (figure 1) et une éruption cutanée diffuse qui n’a pas réagi aux crèmes à base de cortisone. Un bilan infectieux complet s’est révélé négatif.
L’examen physique a révélé des pavillons auriculaires rouges, chauds, enflés et douloureux aux deux oreilles, les lobes étant épargnés (figure 2). Il présentait un gonflement douloureux réparti sur six articulations métacarpophalangiennes et interphalangiennes proximales des deux mains. Il présentait des nodules sous-cutanés rouge vif sur les avant-bras et une éruption maculopapulaire diffuse sur le torse. De légers crépitants étaient audibles à la base des deux poumons à la fin de l’inspiration.
Les examens préliminaires ont révélé une pancytopénie avec une hémoglobine de 115 g/L (volume corpusculaire moyen de 114), des plaquettes de 163x109/L et des leucocytes de 4,1x109/L. Il présentait une élévation de la protéine C réactive (CRP) à 48 mg/l et une vitesse de sédimentation des érythrocytes (VSE) à 43 mm/h, ainsi que des résultats négatifs pour le facteur rhumatoïde, les anticorps antinucléaires et les anticorps anti-cytoplasme des polynucléaires neutrophiles. La biopsie des nodules sous-cutanés a révélé une vascularite leucocytoclasique avec infiltration neutrophile.
L’inflammation multisystémique sur fond de pancytopénie et de SMD chez un homme âgé a fait suspecter un syndrome VEXAS (Vacuoles, Enzyme E1, X-linked, Autoinflammatory, Somatic). L’examen des aspirats de moelle osseuse précédents avec l’hématopathologiste a révélé la présence de vacuoles dans les cellules myéloïdes (figure 3). Un test monogénique a révélé une mutation somatique (p. Met41Leu) dans l’UBA1 à une fraction d’allèle de 73 %, confirmant le diagnostic de VEXAS.
|
Introduction
En 2020, le premier rapport sur le syndrome VEXAS a représenté un changement de paradigme dans notre compréhension des troubles inflammatoires systémiques somatiques acquis à l’âge adulte1. Cette maladie est causée par des variantes du gène UBA1 lié à l’X, qui code une des principales enzymes activatrices d’ubiquitine E1 du corps humain. Comme dans le cas de notre patient, la plupart des variantes pathogènes sont dues à des mutations faux-sens de l’acide aminé méthionine en position 41.
Épidémiologie
L’analyse de l’exome d’environ 163 000 personnes dans une cohorte basée sur un système de santé électronique a révélé que près d’un patient sur 13 000 possédait des variantes UBA1, et que toutes ces personnes présentaient rétrospectivement des caractéristiques compatibles avec le syndrome VEXAS2. Les auteurs notent que cette fréquence est similaire à la prévalence estimée de la maladie de Behçet (1 sur 10 000) et supérieure à d’autres diagnostics rhumatologiques comme la granulomatose avec polyangéite (1 sur 18 000) et la polyartérite noueuse (1 sur 33 000). En tant que maladie acquise liée au chromosome X, elle est principalement observée chez les hommes âgés de plus de 50 ans, mais a également été observée chez les femmes dans de rares cas2-4.
Caractéristiques cliniques
L’identification du syndrome VEXAS se heurte à l’hétérogénéité marquée de sa présentation, qui peut imiter d’autres affections rhumatologiques, notamment la polychondrite récidivante, le syndrome de Sweet, la polyartérite noueuse, la vascularite des petits vaisseaux, la polyarthrite rhumatoïde, la spondylarthrite séronégative et la maladie de Behçet5,6.
Les caractéristiques habituelles comprennent des symptômes constitutionnels, tels que la fièvre, la fatigue et la perte de poids. L’atteinte cutanée est fréquente et variée, comprenant des dermatoses neutrophiliques, des vascularites ou des lésions nodulaires. Une atteinte pulmonaire (infiltrats, épanchements pleuraux), une atteinte oculaire (épisclérite, uvéite, sclérite) ou une arthrite/arthralgie peuvent également survenir chez près de la moitié des patients. Une proportion significative de patients présente également une chondrite nasale ou auriculaire. Une thrombose artérielle ou veineuse non provoquée survient chez environ le tiers des patients. Une lymphadénopathie peut être présente dans les régions hilaire, médiastinale, cervicale, axillaire et inguinale. Les organes moins fréquemment touchés sont le cœur (péricardite, myocardite), le tractus gastro-intestinal (douleurs abdominales, diarrhée, saignements), les reins (protéinurie, hématurie microscopique) et le système nerveux (neuropathie sensorielle, mononévrite multiplexe).
Les analyses sanguines révèlent généralement des cytopénies, notamment une anémie macrocytaire, une lymphopénie, une monocytopénie ou une thrombocytopénie. Au moins 50 % des patients présentent également des caractéristiques du syndrome myélodysplasique, avec ou sans gammapathie monoclonale. La VSE et la CRP sont généralement élevées.
Diagnostic
Le VEXAS doit être envisagé chez les hommes âgés présentant une inflammation généralisée dans le contexte d’une anémie macrocytaire et/ou d’une thrombocytopénie. L’observation de vacuoles cytoplasmiques dans les précurseurs myéloïdes et érythroïdes provenant d’aspirats de moelle osseuse de ces personnes est très suggestive du syndrome VEXAS. Un diagnostic définitif peut être établi grâce à un test génétique portant sur les variantes du gène UBA1.
Approches thérapeutiques
La prise en charge multidisciplinaire doit être adaptée aux caractéristiques de la maladie du patient. Le traitement de soutien peut inclure la prophylaxie des infections/vaccination et la prophylaxie de la thrombose si nécessaire. Des corticostéroïdes à forte dose sont souvent nécessaires, bien que les poussées inflammatoires soient fréquentes lors de la diminution du traitement. Les médicaments antirhumatismaux de fond classiques et les inhibiteurs de l’interleukine (IL)-1, de l’IL-6 et des Janus kinases (JAK) ont été utilisés avec un succès variable, bien qu’une grande partie des patients semblent devenir réfractaires au fil du temps7. Les cytopénies peuvent être traitées par des agents stimulant l’érythropoïétine et/ou l’eltrombopag. L’azacytidine, un agent hypométhylant, s’est également révélé prometteur pour les cytopénies et l’inflammation chez certains patients8,9. La greffe de cellules souches hématopoïétiques (GCSH) allogéniques peut être une alternative de traitement pour les patients atteints d’une maladie grave ou réfractaire au traitement, bien que l’aptitude à la greffe puisse être limitée par l’âge et l’état de santé général du patient, en particulier s’il y a eu une atteinte importante d’un organe majeur liée au VEXAS. Un diagnostic précoce est donc crucial pour limiter l’inflammation et permettre la possibilité d’une greffe de cellules souches avant l’apparition de lésions organiques étendues.
Conclusion
Le syndrome VEXAS doit être envisagé chez les adultes présentant des symptômes constitutionnels inexpliqués, des cytopénies et une inflammation multisystémique pouvant imiter de nombreux autres diagnostics rhumatologiques. Malheureusement, les taux de mortalité demeurent élevés, avec un taux de survie à 5 ans de 63 %6. Des travaux supplémentaires sont nécessaires pour mettre au point des algorithmes de traitement efficaces et fondés sur des données probantes pour cette nouvelle pathologie.
Retour au cas
Le patient a été rapidement traité sous un régime dégressif de prednisone 40 mg par jour et de méthotrexate 25 mg par semaine, par voie sous-cutanée, en tant qu’agent d’épargne stéroïdienne. Il a connu une résolution complète de la chondrite auriculaire et une réponse partielle de l’arthrite, des nodules et de l’éruption cutanée diffuse. La CRP est tombée à 24 mg/L et la VSE à 20 mm/h. La numération cellulaire est restée faible. Un mois plus tard, les manifestations inflammatoires sont réapparues après une réduction progressive des corticostéroïdes à moins de 20 mg. Le ruxolitinib a été envisagé, mais n’a finalement pas été accessible en raison des limitations de l’assurance. Après des discussions avec l’hématologie, l’azacytidine a été mise en place, ce qui a permis de réduire la dose de prednisone à 10 mg par jour. Six mois plus tard, cependant, son SMD a progressé et il est devenu dépendant des transfusions. Il s’est présenté aux urgences avec une dyspnée et une hypoxie et a été admis aux soins intensifs pour une insuffisance respiratoire attribuable à une possible pneumonie. Malgré l’arrêt de tous les immunosuppresseurs et l’administration d’antibiotiques et de vasopresseurs par voie intraveineuse, le patient a développé une défaillance multiorganique et est décédé.
Jason W. An, M.D., M. Sc., FRCPC
Professeur adjoint, Division de rhumatologie,
The Hospital for Sick Children, Toronto (Ontario)
Dilan Dissanayake, M.D., Ph. D., FRCPC
Clinicien-chercheur de transition,
Division de rhumatologie, The Hospital for Sick Children,
Professeur adjoint, Département de pédiatrie,
Université de Toronto, Toronto (Ontario)
Références :
1. Beck DB, Ferrada MA, Sikora KA, et coll. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. Dec 31 2020;383(27):2628-2638. doi:10.1056/NEJMoa2026834
2. Beck DB, Bodian DL, Shah V, et coll. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA. Jan 24 2023;329(4):318-324. doi:10.1001/jama.2022.24836
3. Arlet JB, Terrier B, Kosmider O. Mutant UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. Jun 3 2021;384(22):2163. doi:10.1056/NEJMc2102124
4. Tsuchida N, Kunishita Y, Uchiyama Y, et coll. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis. Aug 2021;80(8):1057-1061. doi:10.1136/annrheumdis-2021-220089
5. Vitale A, Caggiano V, Bimonte A, et coll. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Intern Emerg Med. Apr 2023;18(3):711-722. doi:10.1007/s11739-023-03193-z
6. Georgin-Lavialle S, Terrier B, Guedon AF, et coll. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. Mar 2022;186(3):564-574. doi:10.1111/bjd.20805
7. van der Made CI, Potjewijd J, Hoogstins A, et coll. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunol. Jan 2022;149(1):432-439 e4. doi:10.1016/j.jaci.2021.05.014
8. Comont T, Heiblig M, Riviere E, et coll. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. Feb 2022;196(4):969-974. doi:10.1111/bjh.17893
9. Sockel K, Gotze K, Ganster C, et coll. VEXAS syndrome: complete molecular remission after hypomethylating therapy. Ann Hematol. Jan 12 2024;doi:10.1007/s00277-023-05611-w
|




