Automne 2024 (Volume 34, numéro 3)
Plus qu'une simple affaire de
peau :
voir plus loin que l'épiderme
Par Nadia Luca, M.D., FRCPC, M. Sc.
Télécharger la version PDF
|
Étude de cas
|
| Une fillette de 4 ans, originaire de Jordanie, a été orientée vers la clinique de rhumatologie pédiatrique avec une histoire d’épaississement et de resserrement progressifs de la peau, de raideur articulaire généralisée et de gonflement douloureux bilatéral des mains et des pieds depuis 2 mois. Quelques semaines avant l’apparition des symptômes, elle a eu de la fièvre, de la diarrhée et des ulcères buccaux, et avait été traitée avec de l’amoxicilline. Ses antécédents sont significatifs : retard du langage expressif, polydactylie des mains et des pieds et syndactylie des orteils. Elle ne prenait pas de médicaments sur une base régulière. À l’examen physique, elle présentait une induration diffuse de la peau, plus prononcée sur les extrémités que sur le tronc, le visage étant épargné. Elle présentait également une lésion hyperpigmentée linéaire s’étendant sur la partie postérieure du membre inférieur droit (Figure 1). Elle ne présentait ni dépressions digitales ni ulcères, ni télangiectasies ni capillaires anormaux au niveau des cuticules. Cependant, elle présentait un gonflement de plusieurs articulations, y compris les articulations interphalangiennes proximales bilatérales, les articulations métacarpophalangiennes, les coudes, les poignets, les genoux et les chevilles. De nombreuses articulations étaient maintenues dans une contracture fixe, ce qui limitait considérablement la mobilité. Ses tests laboratoire ont révélé une formule sanguine complète normale, à l’exception d’un nombre d’éosinophiles de 0,9 x 109/L (0,0-0,6 x 109/L). La vitesse de sédimentation des érythrocytes (ESR) était de 62 mm/H (normale < 37 mm/H) et la protéine C-réactive était de 54 mg/L (normale < 10,0 mg/L). La créatine kinase et l’immunoglobuline G étaient élevées à 436 U/L (normale 35-180 U/L) et 19,2 g/L (normale 6,4-14,4 g/L), respectivement. Le facteur rhumatoïde était négatif, l’anticorps anti-nucléaire était positif à un taux de 1:160, et l’anticorps anti-RNP/Sm était également positif. L’imagerie par résonance magnétique (IRM) des extrémités a montré une augmentation du signal T2 dans les fascias sous-cutanés et profonds avec rehaussement au gadolinium. La biopsie du fascia musculaire profond de la jambe inférieure droite a révélé une inflammation périmysiale et endomysiale parcellaire composée de lymphocytes et de plasmocytes sans éosinophiles. Le fascia présentait une inflammation diffuse composée de lymphocytes et de plasmocytes avec une augmentation parcellaire des éosinophiles. |
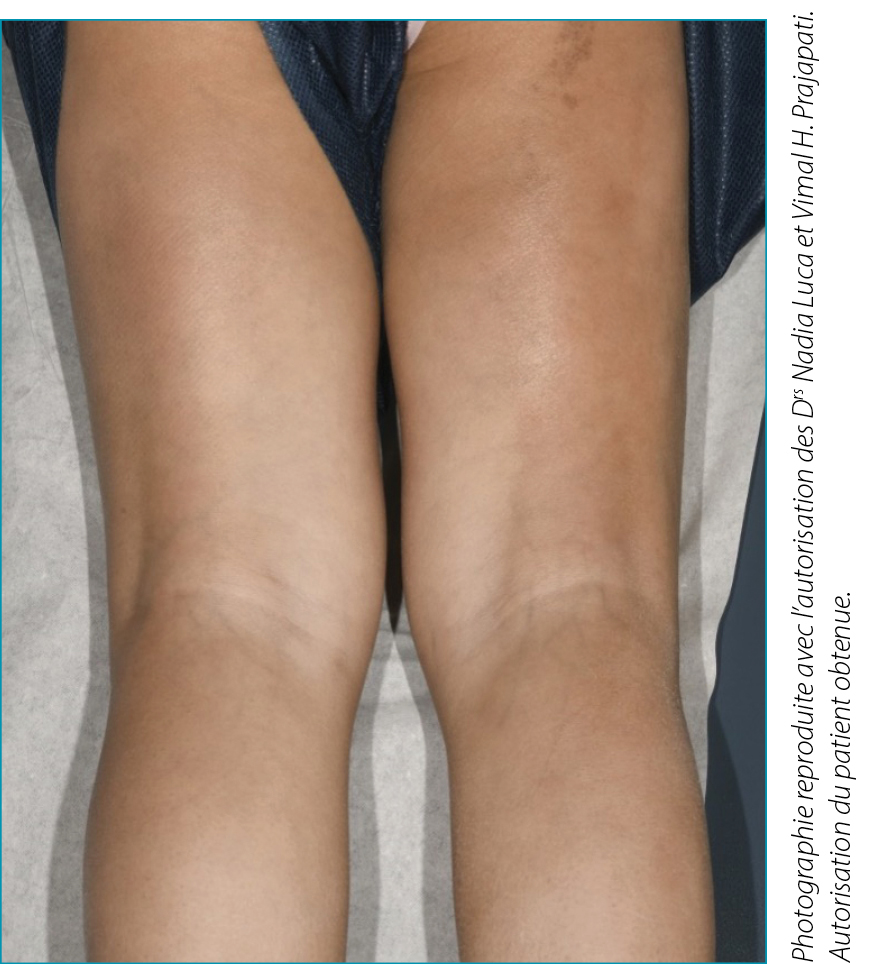
Figure 1. Index patient, montrant une lésion sclérodermique linéaire localisée à la partie postérieure du membre inférieur droit.
Épidémiologie
La fasciite éosinophile (FE) a été reconnue pour la première fois par Shulman, en 1975, comme une maladie diffuse de type sclérodermie caractérisée par une peau ferme et attachée associée à une éosinophilie périphérique et à une hypergammaglobulinémie1. Depuis ce temps, environ 300 cas ont été rapportés et une étude cite une prévalence de 14 par million². La FE affecte principalement les adultes au cours de la quatrième ou de la cinquième décennie, mais tous les âges peuvent être touchés. La plupart des patients sont de race blanche3.
L’étiologie et la pathogénie de la FE restent inconnues. Les déclencheurs potentiels signalés comprennent un effort physique intense ou un traumatisme (davantage chez les adultes que chez les enfants), divers médicaments (par exemple le natalizumab, le vaccin contre la grippe,
la simvastatine, la phénytion, le ramipril), la radiothérapie et Borrelia burgdorferi4. Des maladies auto-immunes et des troubles hématologiques coexistants (par exemple l’anémie aplasique, moins fréquemment des tumeurs malignes) peuvent être présents. Notamment, environ 29 à 50 % des patients atteints de FE présentent simultanément une sclérodermie localisée (SL)5,6. La FE a été décrite tout au long du spectre de la maladie sclérodermique, peut-être à l’extrémité la plus sévère; cependant, son association avec la SL reste à élucider. La plupart des types de SL présentent une sclérose cutanée asymétrique superficielle et discontinue. Cependant, les variantes profondes de la SL peuvent être difficiles à distinguer de la FE. Heureusement, les schémas thérapeutiques pour les deux entités sont assez similaires, ce qui rend la distinction absolue non essentielle.
Caractéristiques cliniques
La FE se caractérise par un œdème symétrique et douloureux des extrémités, suivi d’une sclérose progressive du derme moyen et profond, de la graisse sous-cutanée et du fascia. L’épiderme et le derme superficiel sont largement épargnés. L’aspect caractéristique de la peau d’orange résulte de l’attachement du derme profond aux couches aponévrotiques et musculaires. Le signe du sillon est caractérisé par des dépressions linéaires traversant le trajet des veines superficielles et est mieux visible avec l’élévation du membre (Figure 2). Outre les caractéristiques cutanées, jusqu’à 50 % des patients atteints de la FE peuvent également développer une arthrite inflammatoire et parfois érosive, touchant le plus souvent les mains, les genoux et les poignets. L’induration progressive de la FE peut entraîner des contractures articulaires (par exemple, le billot de prière dans 50 à 67 % des cas), une diminution de la mobilité et un coincement des nerfs4. Des symptômes constitutionnels tels que la fatigue, l’arthralgie, la myalgie et la perte de poids peuvent également être présents. L’atteinte des organes internes est généralement absente dans la FE, ce qui la distingue de la sclérodermie systémique.
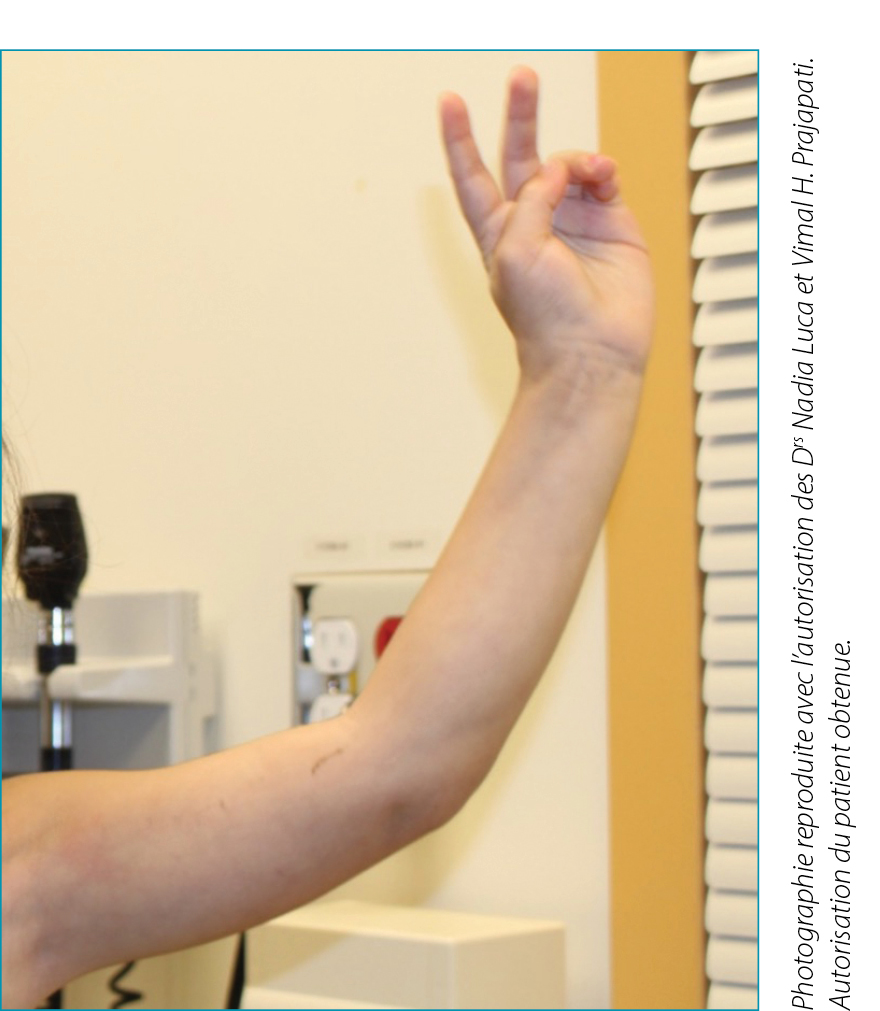
Figure 2. Patient de l’index, présentant un « signe du sillon » au niveau du membre supérieur gauche.
Diagnostic
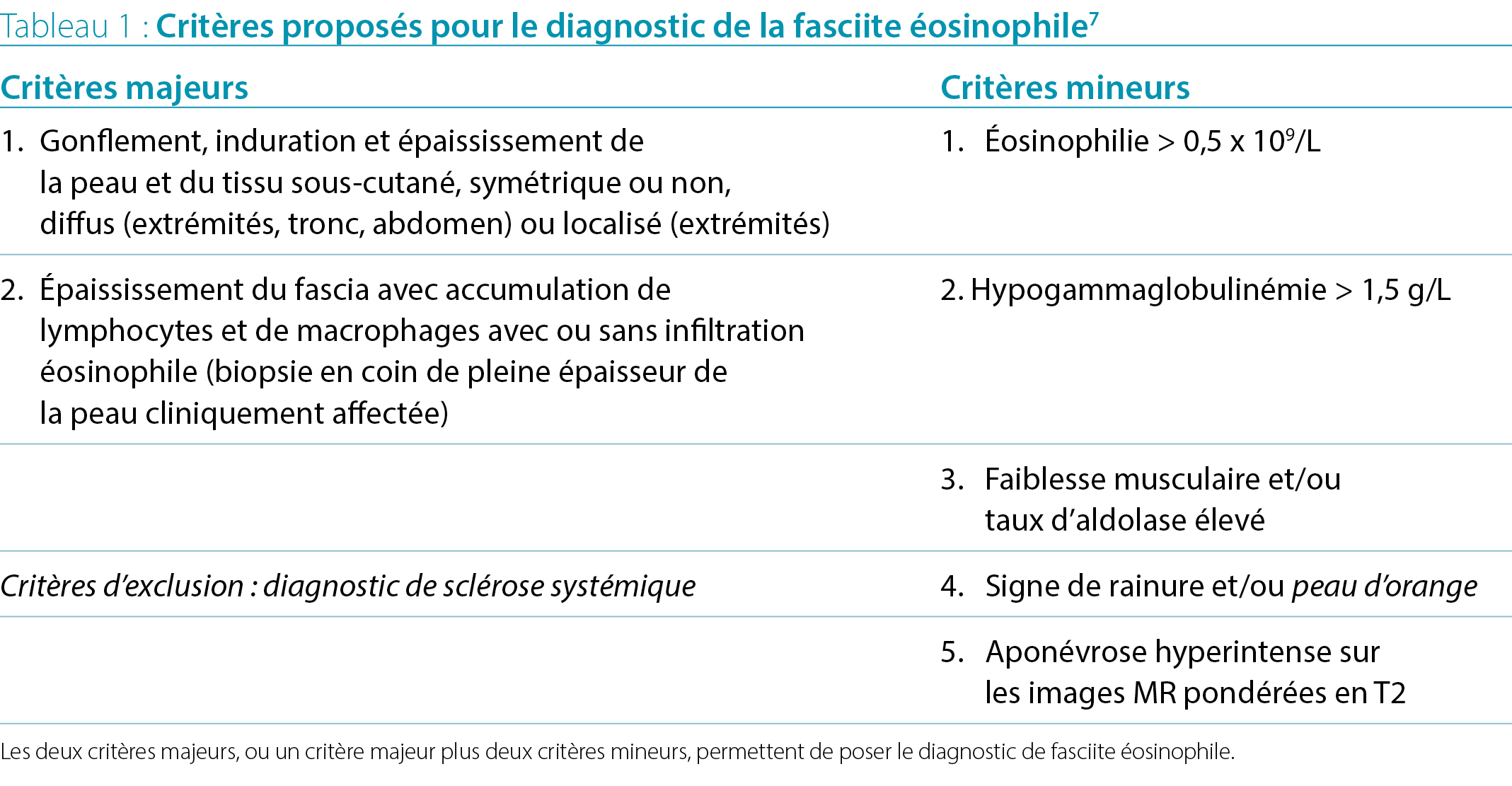
Des critères diagnostiques ont été proposés par Pinal-
Fernandez7, mais n’ont pas été validés (Tableau 1). Le standard de référence pour le diagnostic reste une biopsie en coin de l'épaisseur complète montrant une fascia épaissie, comprenant des lymphocytes et des macrophages, avec ou sans éosinophiles. L’IRM peut identifier un fascia hyperintense sur les images pondérées en T2 et est de plus en plus utilisée pour le diagnostic et le suivi. Les caractéristiques de laboratoire complémentaires comprennent une éosinophilie périphérique présente chez 63 à 93 % des patients (non obligatoire pour le diagnostic), une hypergammaglobulinémie et une ESR élevée. Alors que la biopsie de la peau n’est pas nécessaire dans la majorité des cas de SL, les études histopathologiques d’une biopsie de la peau de pleine épaisseur avec les tissus aponévrotiques et musculaires sont nécessaires pour le diagnostic de la FE.
Approche thérapeutique
Compte tenu de la rareté de la FE, il n’existe pas d’études contrôlées à répartition aléatoire concernant le traitement, et les recommandations actuelles en matière de traitement sont basées sur des études d’observation. Dans son rapport original, Shulman a fait état d’une réponse clinique de
la fasciite à la prednisone sur une période de 15 mois
chez un patient atteint1, et plusieurs études ultérieures décrivent la FE comme répondant aux stéroïdes. On utilise généralement de la prednisone par voie orale à raison de 1 mg/kg/jour, avec une réduction progressive sur plusieurs semaines ou mois. Des impulsions de méthylprednisolone intraveineuse (MP IV) peuvent être utilisées à l’induction pour les cas les plus graves. Une réponse complète est plus probable avec l’ajout d’un deuxième médicament immunosuppresseur, l’agent préféré étant le méthotrexate à raison de 15 à 25 mg par semaine8. Les autres alternatives comprennent le mycophénolate mofétil (MMF) ou l’hydroxychloroquine. Des succès ont également été rapportés avec l’immunoglobuline IV (IVIG)9. Dans les cas réfractaires de FE, divers agents biologiques ont été essayés, les inhibiteurs de l’IL-6 présentant la plus grande fréquence de cas d’amélioration, suivis par les agents anti-TNFα10. Des cas récents de réponse aux inhibiteurs de la Janus kinase ont été publiés.
Le patient de référence a été traité avec des stéroïdes systémiques (d’abord des impulsions IV MP suivies d’une réduction progressive de la prednisone par voie orale), des IgIV mensuelles et du méthotrexate sous-cutané. Elle a constaté une réduction de la douleur, des gonflements de la peau et des articulations, ainsi qu’une amélioration de la mobilité. Cependant, comme elle présentait toujours un resserrement important de la peau et une contracture dans les extrémités, le MMF a été ajouté. Elle a rencontré des difficultés à suivre le traitement par MMF, et une IRM de contrôle a montré une inflammation fasciale persistante. Par conséquent, le tocilizumab a été essayé.
Après plusieurs mois sans amélioration clinique, le traitement a été remplacé par le tofacitinib, qui a conduit à une amélioration modeste supplémentaire.
Par Nadia Luca, M.D., FRCPC, M. Sc.
Rhumatologue pédiatrique et professeure agrégée,
Université d’Ottawa,
Ottawa (Ontario)
Références :
1. Shulman LE. Diffuse Fasciitis with Hypergammaglobulinemia and Eosinophilia: A New Syndrome?
J Rheumatol. Octobre 1984;11(5):569-70. PMID: 6542592.
2. Spielmann L, Arnaud L, Severac F, et coll. Population-based Prevalence of Eosinophilic Fasciitis (Shulman Syndrome): A Capture-recapture Study. Br J Dermatol. Août 2018;179(2):516-517. doi: 10.1111/bjd.16535. Epub 2018 Jun 7. PMID: 29526049.
3. Bischoff L, Derk CT. Eosinophilic Fasciitis: Demographics, Disease Pattern and Response to Treatment: Report of 12 Cases and Review of the Literature. Int J Dermatology. 2008; 47:29-35. https://doi.org/10.1111/j.1365-4632.2007.03544.x
4. Fett N, Arthur M. Eosinophilic Fasciitis: Current Concepts. Clin Dermatol. 2018 Jul-Aug; 36(4):487-497. doi: 10.1016/j.clindermatol.2018.04.006. Epub 2018 Apr 10. PMID: 30047432.
5. Mazilu D, Boltasiu Tataru LA, et coll. Eosinophilic Fasciitis: Current and Remaining Challenges. Int J Mol Sci. 2023 Janvier 2024;24(3):1982. doi: 10.3390/ijms24031982. PMID: 36768300; PMCID: PMC9916848.
6. Stubbs LA, Ogunbona O, Beil E, et coll. Juvenile Eosinophilic Fasciitis: A Single Center Case Series. Pediatr Rheumatol. 2024; 22(29). https://doi-org.ezproxy.lib.ucalgary.ca/10.1186/s12969-024-00960-w.
7. Pinal-Fernandez I, Selva-O'Callaghan A, Grau JM. Diagnosis and Classification of Eosinophilic Fasciitis. Autoimmun Rev. 2014 Apr-May; 13(4-5):379-82. doi: 10.1016/j.autrev.2014.01.019. Epub 2014 Jan 11. PMID: 24424187.
8. Wright NA, Mazori DR, Patel M, et coll. Epidemiology and Treatment of Eosinophilic Fasciitis: An Analysis of 63 Patients From 3 Tertiary Care Centers. JAMA Dermatol. Janvier 2016;152(1):97-9. doi: 10.1001/jamadermatol.2015.3648. PMID: 26559760.
9. Obiakor B, Fan W, Jacobson R, et coll. Functional and Cutaneous Treatment Outcomes With Intravenous Immunoglobulin for Eosinophilic Fasciitis: A Retrospective Study. J Drugs Dermatol. 1er avril 2024;23(4): e107-e109. doi: 10.36849/JDD.8017. PMID: 38564381.
10. Mufti A, Kashetsky N, Abduelmula A, et coll. Biologic Treatment Outcomes in Refractory Eosinophilic Fasciitis: A Systematic Review of Published Reports. J Am Acad Dermatol. Avril 2022;86(4):951-953. doi: 10.1016/j.jaad.2021.03.089. Epub 2 avril 2021. PMID: 33812957.
|




