Spring 2024 (Volume 34, Number 1)
A Vexing Case of Inflammation
By Jason An, MD, MSc, FRCPC; and
Dilan Dissanayake, MD, PhD, FRCPC
Download PDF
|
Case Presentation
|
A 78-year-old male was referred to rheumatology for suspected relapsing polychondritis. His past medical history was significant for myelodysplastic syndrome (MDS) confirmed on bone marrow biopsy by hematology.
He first noticed redness, pain, and swelling of the nose and right ear four months prior. He visited numerous walk-in clinics and had not responded to multiple courses of antibiotics. He responded to a short course of steroids, however, the symptoms recurred upon tapering.
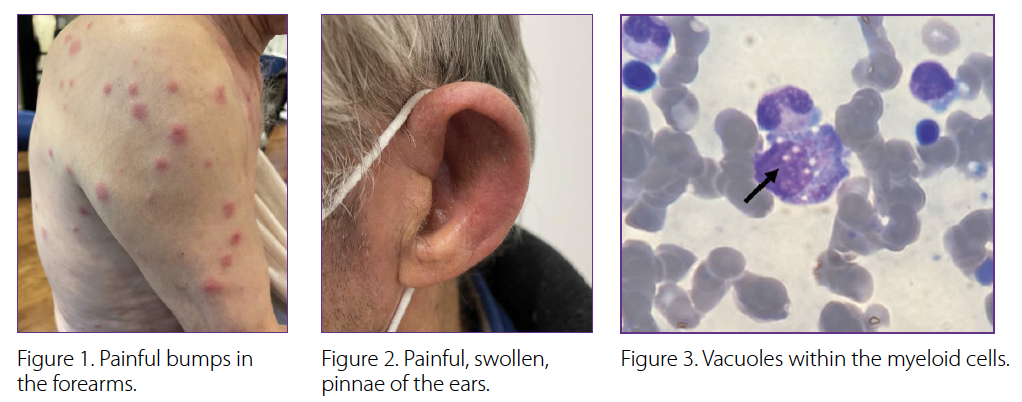
There were also numerous other unexplained symptoms in the preceding months. These included recurrent fevers of up to 39⁰C, a 15-pound unintentional weight loss, bilateral painful red eyes, mouth dryness, and pain in multiple joints of the hands. He noticed painful bumps in the forearms (Figure 1), and a diffuse rash that did not respond to cortisone creams. A thorough infection work-up was negative.
His physical exam revealed red, warm, swollen, and painful pinnae of both ears with sparing of the lobes (Figure 2). There was painful swelling distributed across six metacarpophalangeal and proximal interphalangeal joints of both hands. There were bright red subcutaneous nodules over the forearms and diffuse maculopapular rash over the torso. Faint inspiratory crackles were audible at both lung bases.
Preliminary investigations revealed pancytopenia with hemoglobin 115 g/L (mean corpuscular volume 114), platelets 163x109/L, and leukocytes 4.1x109/L. He had elevated C-reactive protein (CRP) of 48 mg/L and erythrocyte sedimentation rate (ESR) of 43 mm/hr, and negative rheumatoid factor, antinuclear antibodies, and anti-neutrophil cytoplasmic antibodies. Biopsy of the subcutaneous nodules revealed leukocytoclastic vasculitis with neutrophilic infiltration.
The multisystem inflammation on a background of pancytopenia and MDS in an elderly male prompted suspicion of VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. Review of previous bone marrow aspirates with the hematopathologist revealed vacuoles within the myeloid cells (Figure 3). Single gene testing was significant for a somatic mutation (p.Met41Leu) in UBA1 at an allele fraction of 73%, confirming the diagnosis of VEXAS. |
Introduction
In 2020, the first report of VEXAS syndrome represented a paradigm shift in our understanding of somatically acquired adult-onset systemic inflammatory disorders.1 The condition is caused by variants in the X-linked UBA1 gene, which encodes one of the main E1 ubiquitin-activating enzymes of the human body. As with our patient, most pathogenic variants are due to missense changes of the amino acid methionine at position 41.
Epidemiology
Exome analysis of approximately 163,000 individuals in an electronic-health-system-based cohort revealed that nearly 1 in 13,000 patients possessed UBA1 variants, with all of these individuals retrospectively found to have features consistent with VEXAS syndrome.2 The authors noted that this frequency is similar to the estimated prevalence of Behçet disease (1 in 10,000) and higher than other rheumatologic diagnoses such as granulomatosis with polyangiitis (1 in 18,000) and polyarteritis nodosa (1 in 33,000). As an acquired X-linked disease, it is primarily observed in males over the age of 50 years but has also been described in females in rare cases.2-4
Clinical Features
A challenge in identifying VEXAS syndrome is the marked heterogeneity in its presentation, which can mimic other rheumatologic conditions, including relapsing polychondritis, Sweet syndrome, polyarteritis nodosa, small vessel vasculitis, rheumatoid arthritis, seronegative spondyloarthritis and Behçet disease.5,6
Typical features include constitutional symptoms, such as fever, fatigue, and weight loss. Skin involvement is common and varied, including neutrophilic dermatoses, vasculitis, or nodular lesions. Pulmonary involvement (infiltrates, pleural effusions), ocular involvement (episcleritis, uveitis, scleritis) and/or arthritis/arthralgia can also occur in up to half of patients. A significant proportion of patients also present with nasal or auricular chondritis. Unprovoked arterial or venous thrombosis occurs in approximately a third of patients. Lymphadenopathy can be present in the hilar, mediastinal, cervical, axillary and inguinal areas. Less commonly involved organs are the heart (pericarditis, myocarditis), gastrointestinal tract (abdominal pain, diarrhea, bleeding), kidneys (proteinuria, microscopic hematuria), and nervous system (sensory neuropathy, mononeuritis multiplex).
Bloodwork will commonly show cytopenias, including macrocytic anemia, lymphopenia, monocytopenia and/or thrombocytopenia, with at least 50% of patients also having features of myelodysplastic syndrome, with or without monoclonal gammopathy. ESR and CRP are typically elevated.
Diagnosis
VEXAS should be suspected in elderly males presenting with widespread inflammation in the context of macrocytic anemia and/or thrombocytopenia. Identification of cytoplasmic vacuoles in myeloid and erythroid precursors from bone marrow aspirates of such individuals are highly suggestive of VEXAS syndrome. Definitive diagnosis can be reached through genetic testing for variants in the UBA1 gene.
Therapeutic Approaches
Multidisciplinary management should be tailored to the disease features of the patient. Supportive treatment may include infection prophylaxis/vaccination and thrombosis prophylaxis as indicated. High-dose corticosteroids are often required, although inflammatory flares are common upon tapering. Conventional disease-modifying antirheumatic drugs and inhibitors of interleukin (IL)-1, IL-6, and Janus kinases have been used with varying success, though a large proportion of patients appear to become refractory over time.7 Cytopenias may be treated with erythropoietin-stimulating agents and/or eltrombopag. The hypomethylating agent, azacytidine, has also showed promise for both cytopenias and inflammation in certain patients.8,9 Allogeneic hematopoietic stem cell transplantation (HSCT) may be a curative option for patients with severe or treatment-refractory disease, though suitability for transplant can be limited by the age and overall health of the patient, particularly if there has been significant VEXAS-related major organ involvement. Early diagnosis is therefore crucial in order to limit inflammation and allow for the possibility of HSCT before the onset of extensive organ damage.
Back to the Case
The patient was promptly treated with a tapering regimen of prednisone 40mg daily and weekly methotrexate 25 mg subcutaneously as a steroid-sparing agent. He experienced a complete resolution in ear chondritis, and the partial response of arthritis, nodules, and diffuse rash. There were reductions in CRP to 24 mg/L, and ESR to 20 mm/hr. Cell counts remained low. One month later, the inflammatory manifestations recurred following tapering of corticosteroids below 20 mg. Ruxolitinib was considered but ultimately not accessible due to insurance limitations. After discussions with hematology, azacytidine was initiated, which allowed his prednisone to be tapered to 10 mg daily. Six months later, however, his MDS progressed, and he became transfusion-dependent. He presented to the ER with dyspnea and hypoxia and was admitted to intensive care for respiratory failure due to suspected pneumonia. Despite stopping all immunosuppressives, and administering intravenous antibiotics and vasopressors, the patient developed multiorgan failure and died.
Conclusion
VEXAS syndrome can mimic many other rheumatologic diagnoses, and should be considered in older adults with unexplained constitutional symptoms, cytopenias, and multi-system inflammation. Unfortunately, mortality rates remain high with a 5-year survival rate of 63%.6 Further work is required to develop effective evidence-based treatment algorithms for this new condition.
Jason W. An, MD, MSc, FRCPC
Assistant Professor,
Division of Rheumatology,
The Hospital for Sick Children, Toronto, Ontario
Dilan Dissanayake, MD, PhD, FRCPC
Transition Clinician-Scientist,
Division of Rheumatology, The Hospital for Sick Children
Assistant Professor, Department of Pediatrics,
University of Toronto, Toronto, Ontario
References:
1. Beck DB, Ferrada MA, Sikora KA, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. Dec 31 2020;383(27):2628-2638. doi:10.1056/NEJMoa2026834
2. Beck DB, Bodian DL, Shah V, et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA. Jan 24 2023;329(4):318-324. doi:10.1001/jama.2022.24836
3. Arlet JB, Terrier B, Kosmider O. Mutant UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. Jun 3 2021;384(22):2163. doi:10.1056/NEJMc2102124
4. Tsuchida N, Kunishita Y, Uchiyama Y, et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis. Aug 2021;80(8):1057-1061. doi:10.1136/annrheumdis-2021-220089
5. Vitale A, Caggiano V, Bimonte A, et al. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Intern Emerg Med. Apr 2023;18(3):711-722. doi:10.1007/s11739-023-03193-z
6. Georgin-Lavialle S, Terrier B, Guedon AF, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. Mar 2022;186(3):564-574. doi:10.1111/bjd.20805
7. van der Made CI, Potjewijd J, Hoogstins A, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunol. Jan 2022;149(1):432-439 e4. doi:10.1016/j.jaci.2021.05.014
8. Comont T, Heiblig M, Riviere E, et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. Feb 2022;196(4):969-974. doi:10.1111/bjh.17893
9. Sockel K, Gotze K, Ganster C, et al. VEXAS syndrome: complete molecular remission after hypomethylating therapy. Ann Hematol. Jan 12 2024;doi:10.1007/s00277-023-05611-w
|




