Fall 2024 (Volume 34, Number 3)
More Than Skin Deep:
Thinking Below the Surface
By Nadia Luca, MD, FRCPC, MSc
Download PDF
|
Case Presentation
|
| A 4-year-old girl originally from Jordan was referred to the pediatric rheumatology clinic with a 2-month history of progressive thickening and tightening of her skin, generalized joint stiffness and painful bilateral swelling of her hands and feet. A few weeks prior to onset of symptoms, she had fever, diarrhea and oral ulcers and was treated with amoxicillin. Her history was significant for expressive language delay, polydactyly of the hands and feet, and syndactyly of the toes. She took no regular medications. On physical examination she had diffuse induration of the skin, more pronounced on the extremities than the trunk, with sparing of her face. She also had a linear hyperpigmented lesion extending down the posterior right lower extremity (Figure 1). She did not have any digital pitting or ulcers, telangiectasia or abnormal nailfold capillaries. She had swelling of several joints, including bilateral proximal interphalangeal joints, metacarpophalangeal joints, elbows, wrists, knees, and ankles. Many of the joints were held in fixed contracture resulting in very limited mobility. Her laboratory testing revealed a normal complete blood count apart from eosinophils of 0.9 x 109/L (0.0-0.6 x 109/L). Erythrocyte sedimentation rate (ESR) was 62 mm/Hr (normal <37 mm/Hr) and C-reactive protein was 54 mg/L (normal <10.0 mg/L). Creatine kinase and immunoglobulin G were elevated at 436 U/L (normal 35-180 U/L) and 19.2 g/L (normal 6.4-14.4 g/L), respectively. Rheumatoid factor was negative, anti-nuclear antibody was positive at a titre of 1:160, and anti-RNP/Sm antibody was also positive. Magnetic resonance imaging (MRI) of the extremities demonstrated increased T2 signal in the subcutaneous and deep fascia with gadolinium enhancement. Biopsy of the deep muscular fascia of the right lower leg revealed patchy perimysial and endomysial inflammation composed of lymphocytes and plasma cells with no eosinophils. The fascia showed diffuse inflammation composed of lymphocytes and plasma cells with a patchy increase in eosinophils. |
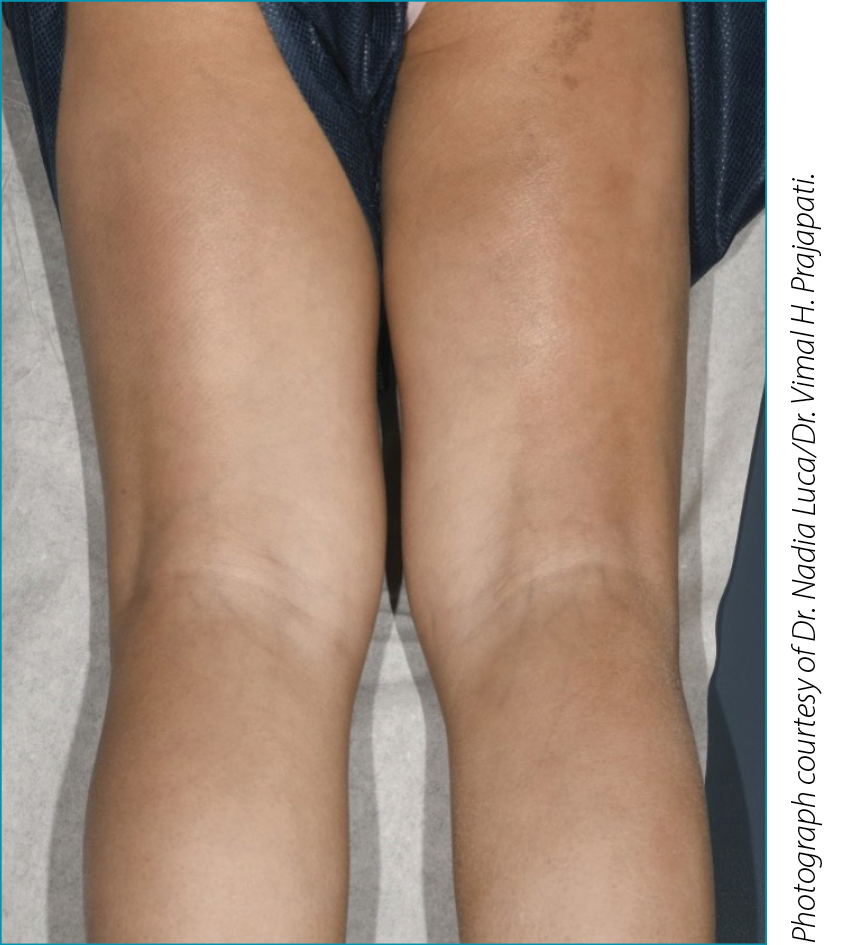
Figure 1. Index patient, demonstrating linear localized
scleroderma lesion at posterior right lower limb.
Epidemiology
Eosinophilic fasciitis (EF) was first recognized by Shulman in 1975 as a diffuse scleroderma-like illness characterized by firm bound-down skin associated with peripheral eosinophilia and hypergammaglobulinemia.1 Since then, approximately 300 cases have been reported, and one study cites a prevalence of 14 per million.2 EF primarily affects adults in the fourth or fifth decades, but all ages may be affected. Most reported patients are Caucasian.3
The etiology and pathogenesis of EF remain unknown. Reported potential triggers include intense physical exertion or trauma (more so in adult versus pediatric patients), various drugs (e.g. natalizumab, influenza vaccine, simvastatin, phenytoin, ramipril), radiation therapy, and Borrelia burgdorferi infection.4 Co-existing autoimmune diseases and hematologic disorders (e.g. aplastic anemia, less frequently malignancies) may be present. Notably, approximately 29-50% of patients with EF simultaneously present with localized scleroderma (LS).5,6 EF has been described along the spectrum of scleroderma-
like disease, perhaps at the more severe end; however, its association with LS remains to be elucidated. Most types of LS have superficial, discontinuous asymmetric cutaneous sclerosis. However, deep variants of LS may be difficult to distinguish from EF. Fortunately, the treatment regimens for both entities are quite similar, making absolute distinction nonessential.
Clinical Features
EF is characterized by symmetrical, painful edema of the extremities followed by progressive sclerosis of the mid and deep dermis, subcutaneous fat and fascia. The epidermis and superficial dermis are largely spared. The characteristic peau d’orange appearance occurs as a result of the deep dermis becoming tethered to the fascial and muscle layers. The groove sign is characterized by linear depressions traversing along the course of superficial veins and is best seen with limb elevation (Figure 2). In addition to the characteristic cutaneous features, up to 50% of patients with EF may also develop an inflammatory and occasionally erosive arthritis, most commonly involving the hands, knees and wrists. Progressive induration in EF can lead to joint contractures (e.g. prayer sign in 50-67% of cases), decreased mobility and nerve entrapment.4 Constitutional symptoms such as fatigue, arthralgia, myalgia and weight loss may also be present. Internal organ involvement is generally absent in EF, distinguishing it from systemic scleroderma.
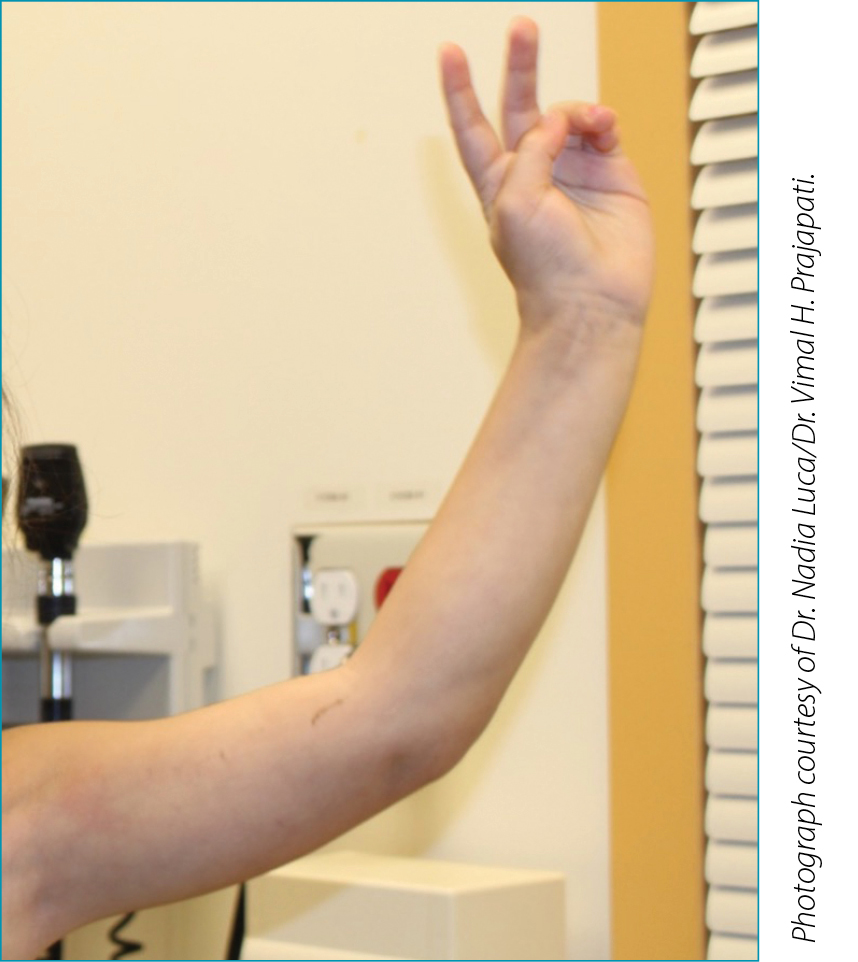
Figure 2. Index patient, demonstrating “groove sign”
in left upper extremity.
Diagnosis
Diagnostic criteria have been proposed by Pinal-Fernandez7 but have not been validated (Table 1). The gold standard for diagnosis remains a full-thickness wedge biopsy demonstrating thickened fascia including lymphocytes and macrophages, with or without eosinophils. MRI may identify hyperintense fascia on T2-weighted images and is increasingly used for diagnosis and monitoring. Supportive laboratory features include peripheral eosinophilia present in 63-93% of patients (not mandatory for diagnosis), hypergammaglobulinemia and elevated ESR. While skin biopsy is not necessary in a majority of LS cases, histopathologic studies of a full-thickness skin biopsy with fascia and muscle tissues are required for the diagnosis of EF.

Therapeutic Approach
Given the rarity of EF, there are no randomized controlled studies regarding treatment, and current treatment recommendations are based on observational studies. In his original report, Shulman reported clinical response of fasciitis to prednisone therapy over 15 months in one affected patient1, and several subsequent studies describe EF as steroid-responsive. Oral prednisone at a dose of 1 mg/kg/day tapered over weeks or months is typically used. Intravenous methylprednisolone (IV MP) pulses may be used at induction for more severe cases. Complete response is more likely with the addition of a second immunosuppressive drug, the preferred agent being methotrexate 15-25 mg weekly.8 Other alternatives include mycophenolate mofetil (MMF) or hydroxychloroquine. Success has also been reported with IV immunoglobulin (IVIG).9 In refractory cases of EF, various biologic agents have been tried, with IL-6 inhibitors having the greatest frequency of cases with improvement, followed by anti-TNFα agents.10 Recent case reports of response to Janus kinase inhibitors have been published.
The index patient was treated with systemic steroids (initially IV MP pulses followed by oral prednisone taper), monthly IVIG, and subcutaneous methotrexate. She experienced reduction in pain, skin and joint swelling and improved mobility. However, as she still had significant tightening of skin and contracture in the extremities, MMF was added. She had difficulty with adherence to MMF and repeat MRI demonstrated some ongoing fascial inflammation, thus tocilizumab was tried. After several months there was no clinical improvement and she was switched to tofacitinib, which resulted in modest additional improvement.
Nadia Luca, MD, FRCPC, MSc
Pediatric Rheumatologist and Associate Professor,
University of Ottawa
Ottawa, Ontario
References:
1. Shulman LE. Diffuse Fasciitis with Hypergammaglobulinemia and Eosinophilia: A New Syndrome?
J Rheumatol. 1984 Oct; 11(5):569-70. PMID: 6542592.
2. Spielmann L, Arnaud L, Severac F, et al. Population-based Prevalence of Eosinophilic Fasciitis (Shulman Syndrome): A Capture-recapture Study. Br J Dermatol. 2018 Aug; 179(2):516-517. doi: 10.1111/bjd.16535. Epub 2018 Jun 7. PMID: 29526049.
3. Bischoff L, Derk CT. Eosinophilic Fasciitis: Demographics, Disease Pattern and Response to Treatment: Report of 12 Cases and Review of the Literature. Int J Dermatology. 2008; 47:29-35. https://doi.org/10.1111/j.1365-4632.2007.03544.x
4. Fett N, Arthur M. Eosinophilic Fasciitis: Current Concepts. Clin Dermatol. 2018 Jul-Aug; 36(4):487-497. doi: 10.1016/j.clindermatol.2018.04.006. Epub 2018 Apr 10. PMID: 30047432.
5. Mazilu D, Boltasiu Tataru LA, et al. Eosinophilic Fasciitis: Current and Remaining Challenges. Int J Mol Sci. 2023 Jan 19; 24(3):1982. doi: 10.3390/ijms24031982. PMID: 36768300; PMCID: PMC9916848.
6. Stubbs LA, Ogunbona O, Beil E, et al. Juvenile Eosinophilic Fasciitis: A Single Center Case Series. Pediatr Rheumatol. 2024; 22(29). https://doi-org.ezproxy.lib.ucalgary.ca/10.1186/s12969-024-00960-w.
7. Pinal-Fernandez I, Selva-O'Callaghan A, Grau JM. Diagnosis and Classification of Eosinophilic Fasciitis. Autoimmun Rev. 2014 Apr-May; 13(4-5):379-82. doi: 10.1016/j.autrev.2014.01.019. Epub 2014 Jan 11. PMID: 24424187.
8. Wright NA, Mazori DR, Patel M, et al. Epidemiology and Treatment of Eosinophilic Fasciitis: An Analysis of 63 Patients From 3 Tertiary Care Centers. JAMA Dermatol. 2016 Jan; 152(1):97-9. doi: 10.1001/jamadermatol.2015.3648. PMID: 26559760.
9. Obiakor B, Fan W, Jacobson R, et al. Functional and Cutaneous Treatment Outcomes With Intravenous Immunoglobulin for Eosinophilic Fasciitis: A Retrospective Study. J Drugs Dermatol. 2024 Apr 1; 23(4):e107-e109. doi: 10.36849/JDD.8017. PMID: 38564381.
10. Mufti A, Kashetsky N, Abduelmula A, et al. Biologic Treatment Outcomes in Refractory Eosinophilic Fasciitis: A Systematic Review of Published Reports. J Am Acad Dermatol. 2022 Apr; 86(4):951-953. doi: 10.1016/j.jaad.2021.03.089. Epub 2021 Apr 2. PMID: 33812957.
|




