Hiver 2016 (volume 26, numéro 4)
L'analyse des résultats d'essais cliniques en rhumatologie
Par Philip Baer, M.D., C.M., FRCPC, FACR; Michael Starr, M.D.; Nigil Haroon, M.D., Ph.D., D.M.
Télécharger le PDF
Introduction
Avec des centaines d’essais cliniques publiés chaque année, la rhumatologie est un domaine qui évolue rapidement. Il est presque impossible de rester au fait de la documentation, et il est presque tout aussi difficile de discerner les essais qui génèrent de précieuses connaissances de ceux qui ont peu à offrir aux rhumatologues praticiens.
Cet article a pour objectif d'aider le lecteur à résoudre ce problème. Ainsi, nous décrirons brièvement les caractéristiques des essais bien et mal conçus, servant de guide au moment d'estimer si le plan, les analyses ou les conclusions d’une étude sont erronés, allant du mauvais choix de patients à l’identification inappropriée d’un effet de classe parmi les agents. Nous avons de plus créé une liste de contrôle, qui peut être utilisée pour évaluer rapidement les nouveaux rapports de recherche et interpréter leurs conclusions.
Le plan de l’étude
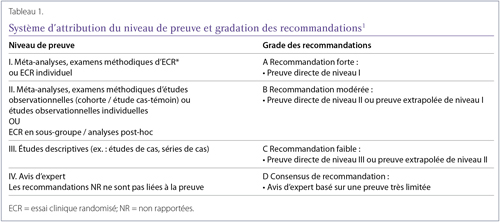
Le risque de biais varie selon le type de preuves cliniques disponibles. Il est important d’être conscient du niveau de la preuve avant d’effectuer l’interprétation des résultats. La gradation des recommandations varie en fonction du niveau de la preuve pour répondre à une question clinique ou une question de recherche donnée. Ce principe est universellement reconnu et a récemment été utilisé dans les lignes directrices sur la polyarthrite rhumatoïde (PR) de la Société canadienne de rhumatologie (Tableau 1).
Type d’étude
Les essais prospectifs attendent et suivent l’élaboration des résultats au fil du temps dans la population choisie. Les essais rétrospectifs examinent les dossiers antérieurs pour déterminer si des facteurs de risque ou de précédentes interventions différant entre deux groupes ont influencé des résultats précis. Même si les essais rétrospectifs peuvent produire des données utiles à long terme, certains types de biais sont plus fréquents dans les études rétrospectives que dans celles prospectives, et cela peut affecter la validité de leurs résultats2,3. De plus, les études rétrospectives peuvent ne pas disposer des paramètres de référence nécessaires devant faire l’objet d’une surveillance pour évaluer les effets indépendants3. Par exemple, un examen rétrospectif de 35 ans portant sur l’arthrite septique en tant que complication de la polyarthrite rhumatoïde (PR) a été réalisé au même établissement, mais l’information sur l’activité de la maladie, les résultats fonctionnels et les dommages structuraux, des facteurs qui pourraient influer sur les résultats, n’étaient pas disponibles pour la plupart des patients4.
La randomisation et l’insu sont d’excellentes méthodes pour réduire les biais au minimum. Si la répartition des patients dans les groupes de traitement n’est pas aléatoire, les investigateurs peuvent, par inadvertance, assigner des patients qui sont davantage malades au groupe de traitement qu’ils estiment être le plus efficace. S’il ne s’agit pas d’une étude à l’insu, les patients, les investigateurs et les évaluateurs des résultats peuvent surestimer les effets des traitements, particulièrement pour les résultats évalués de façon subjective5. Le manque de randomisation et les essais sans insu ont été associés à une augmentation de la probabilité qu’une nouvelle thérapie soit supérieure au comparateur6.
Même les résultats d’essais cliniques randomisés obtenus post hoc ou les analyses des sous-groupes sont sujets à des biais 7,8. Une analyse post hoc examine les données après l’achèvement de l'essai et les rapports sur les critères d’évaluation qui n’ont pas été préétablis dans le plan de l’étude. Il a été suggéré que le fait de ne pas révéler au lecteur qu’une analyse a été effectuée après coup devrait être considéré comme un cas d’inconduite scientifique9.
Les analyses des sous-groupes consistent en l'analyse de données de groupes de patients précis (répartis selon l’âge, le sexe, la gravité de la maladie ou tout autre facteur) afin de déterminer si un traitement a notamment bien fonctionné pour un type de patient en particulier. Malheureusement, si suffisamment de sous-groupes sont spécifiés, la probabilité d’avoir un résultat faussement positif augmente : effectuer l'analyse des résultats de 10 sous-groupes entraîne une probabilité qu’il y en ait au moins un qui produise un résultat faussement positif statistiquement significatif (p < 0,05)7. Un facteur de correction pour les comparaisons multiples, comme la correction de Bonferroni, devrait être utilisé une telle situation10.
La population de patients
Les cliniciens peuvent appliquer les résultats d’un essai pour traiter un patient seulement si les sujets participant à l’essai sont semblables à ceux vus dans la pratique clinique. Le premier tableau d’un rapport d’essai clinique résume habituellement les caractéristiques de la population de patients, incluant l’âge, le sexe, la gravité ou la durée de la maladie, les comorbidités et l’histoire pharmacothérapeutique. Idéalement, les sujets de l’essai devraient être semblables aux patients vus dans la pratique clinique en ce qui concerne la plupart de ces points. En particulier, les résultats d’essais observés chez les patients qui sont plus ou moins malades, qui appartiennent à un groupe différent, qui sont de sexe différent, qui ont plus ou moins de comorbidités ou dont les traitements antérieurs ont plus ou moins échoué, sont moins susceptibles de s’appliquer à tous les patients souffrant de la maladie à l’étude. Il faut vérifier si des traitements de sauvetage et la prise de médicaments concomitants ont été autorisés et s’il existe des différences entre les mesures de base des groupes de traitement. On estime que seulement 5 % des patients d'une pratique clinique de rhumatologie typique seraient admissibles à des essais cliniques sur la polyarthrite rhumatoïde basés sur des critères courants d’inclusion et d’exclusion11.
Les décisions entourant les techniques de recrutement des patients peuvent donner lieu à une population de patients biaisée. Par exemple, un récent essai sur la douleur lombaire a recruté tous ses patients dans la même clinique de lombalgie dans un hôpital tertiaire12. Une population de patients provenant d’un centre aussi spécialisé peut ne pas refléter la pratique d’un clinicien moyen en matière de facteurs sociodémographiques et de facteurs liés au mode de vie ou aux circonstances, et les résultats de l’essai pourraient ne pas s’appliquer aux patients en dehors de cette population.
Pour éviter cette limitation, les essais à grande échelle recrutent souvent des patients à partir d’un large éventail de centres, idéalement dans différentes régions du monde. Cependant, cela comporte des risques de manque de normalisation de l’intervention et des mesures des résultats. De plus, les essais effectués dans des régions avec un accès insuffisant aux soins de santé peuvent afficher un taux de réponse supérieur à la normale dans le groupe sous placebo, en raison des patients qui demeurent dans l’essai afin de pouvoir accéder à des soins médicaux autrement inaccessibles (P. Baer, communication personnelle, 15 juin 2016). Les profils de risques rapportés dans les études menées dans des populations avec des taux plus élevés de maladies endémiques géographiquement, comme la tuberculose ou l’hépatite, ne s’appliquent pas nécessairement à d’autres populations.
Parfois, un essai est conçu pour inclure seulement les patients d'une certaine catégorie d’âge ou de gravité de maladie. Par exemple, même si l’essai TEMPO comprenait des patients atteints de PR avec tout niveau de gravité de la maladie, si un traitement par ARMM autre que le méthotrexate13 avait échoué, une étude TEMPO de prolongation incluait seulement des patients souffrant d’une maladie modérée14. Bien que ce fait ait été clairement divulgué, cela signifie que les résultats de l’étude ne sont pas nécessairement applicables aux patients souffrant d’une forme légère ou grave de la maladie.
De même, l’essai RAPID-axSpA a étudié le certolizumab chez les patients atteints de spondylarthrite axiale (axSpA) et a révélé un avantage important par rapport au placebo. Cependant, l’étude a seulement recruté des patients avec un taux de protéine C réactive (CRP) au-dessus de 7,9 mg/L ou une sacro-illite sur IRM selon la définition de l’ASAS/OMERACT. Ces résultats ne peuvent donc s’appliquer qu’aux patients présentant ces caractéristiques15.
Les populations à l’étude doivent aussi être suffisamment importantes pour déceler une différence réelle entre les traitements, s’il en existe une. Sinon, un résultat négatif peut perdre de son sens et l’essai peut induire en erreur les cliniciens à la recherche d’information applicable à leur pratique. Le nombre de patients requis pour déceler une différence dépend de la fréquence ou de la variabilité des résultats mesurés et des effets escomptés de la ou des interventions à l’étude, entre autres facteurs. Les statisticiens ont développé des formules (calcul des effets du traitement) pour la calculer16,17. Cependant, parce que le recrutement de patients peut être plus difficile que prévu, les études peuvent être moins puissantes et conclure à tort qu’il n’y a pas de différence entre les groupes de traitement (erreur de type 2). De plus, les essais font rarement des calculs des effets du traitement pour les analyses de sous-groupes, les rendant souvent moins puissants avec un risque encore plus grand d’obtenir des résultats faussement négatifs18.
Il s’agissait jadis d’une question importante pour les essais en rhumatologie19, mais cela peut encore se produire. Par exemple, en 2012, un essai comparatif sur la physiothérapie et l’acupuncture chez les patients souffrant d’arthrose du genou sévère et en attente d’une chirurgie n’a trouvé aucun avantage par rapport aux soins habituels, mais on n’a pas obtenu la taille de l’échantillon requis20. En 2014, un essai comparant l’étanercept plus le méthotrexate à divers ARMM plus le méthotrexate n’a pas montré de différence notable pour certains paramètres en raison de l’attrition de patients, ce qui a mené à une étude moins puissante22.
De la même manière, l’étude ABILITY-1 portant sur la spondylarthrite axiale non radiographique (nraxSpA) excluait les patients qui remplissaient les critères de New York modifiés pour la SpA. Toutefois, une relecture indépendante a posteriori des rayons X de 102 patients a entraîné le reclassement de 38 d'entre eux, faisant en sorte qu’ils remplissaient ces critères. Puisque l’essai n’incluait que 185 patients, cette reclassification s’est traduite par une faible puissance et le Comité consultatif sur l'arthrose (Arthritis Drugs Advisory Committee) de la FDA (États-Unis) a rejeté une demande visant à étendre l’indication de l’adalimumab aux patients atteints de nraxSpA23.
Les interventions
Dans les essais non contrôlés par placebo, le choix d’agents de comparaison est vital : une comparaison avec une intervention moins efficace que la norme de soins exigée pour la maladie traitée ne reflètera pas fidèlement l’utilité clinique de l’agent testé24. L’essai ADACTA est un exemple de la question en cause. Cet essai a montré que le tocilizumab en monothérapie était supérieur à l’adalimumab en monothérapie chez les patients atteints de PR. L’adalimumab a été choisi parce qu’il était « un traitement biologique de première ligne adopté à l’échelle mondiale (utilisé conjointement avec le méthotrexate et en monothérapie) chez les patients souffrant de PR qui sont réfractaires aux médicaments ARMM non biologiques25 ». Cependant, l’efficacité connue du tocilizumab comme agent administré en monothérapie, par rapport à l’utilisation habituelle de l’adalimumab conjointement avec le méthotrexate, doit être prise en compte au moment d’examiner les résultats de l’essai.
Pour être utiles dans la pratique clinique, les essais doivent aussi refléter les doses couramment utilisées/approuvées du médicament testé et de son ou ses médicaments de comparaison. Par exemple, un essai sur la polyarthrite psoriasique a comparé l’utilisation hebdomadaire de 7,5 à 15 mg de méthotrexate par rapport au placebo pour déterminer l’efficacité et l’innocuité de faibles doses de méthotrexate. L’essai n’a décelé aucune différence significative dans le nombre d’articulations enflées et douloureuses avec l’utilisation de méthotrexate. Toutefois, étant donné que la dose sélectionnée était sensiblement inférieure à celle utilisée en pratique clinique, les résultats de l’essai ne sont pas réellement applicables à la pratique des cliniciens canadiens26.
De même, l’essai sur la douleur lombaire a comparé l’administration d’une dose de 400 mg/jour (la dose maximale quotidienne recommandée) de célécoxib à 1 000 mg/jour d’acétaminophène (la dose maximale quotidienne recommandée est de 3 200 à 4 000 mg/jour)12. Il n’est pas surprenant que le célécoxib ait démontré des effets supérieurs sur la douleur. Un autre exemple est l’essai SATORI, portant sur la PR, qui a comparé l’administration d’une dose de 8 mg/semaine de tocilizumab avec du méthotrexate, une dose bien inférieure à celle habituellement utilisée en Amérique du Nord27.
Il est à noter que même les essais contrôlés par placebo peuvent avoir un biais inhérent, puisque la voie d’administration du médicament a le potentiel d'influencer la perception qu'ont les participants de l’efficacité d’une intervention28,29. Dans la mesure du possible, les essais bien conçus veilleront à ce que la même voie d'administration soit utilisée pour tous les groupes de traitement, y compris celui sous placebo.
Les critères d’évaluation
Pour que l’essai produise des résultats valides, les critères d’évaluation de l’étude doivent être définis avec soin. Le DAS (Disease Activity Score), les réponses de l'American College of Rheumatology (ACR) et les résultats des analyses de sang ont des critères préétablis qui améliorent leur reproductibilité, mais des critères subjectifs comme l’évaluation des limitations fonctionnelles, l’activité de la maladie ou la qualité de vie peuvent être plus vagues et peuvent faire l’objet de désaccords. Des critères de substitution, comme les niveaux de marqueurs inflammatoires, peuvent être faussement qualifiés de résultats de la maladie même lorsqu’ils sont moins significatifs que les résultats principaux, comme la rémission. Les critères non validés (comme l’utilisation de mesures de spondylarthrite, tel le système de pointage du Consortium canadien de recherche sur la spondylarthrite [CCRS] dans une étude sur la douleur lombaire de nature mécanique12) peuvent ne pas corréler avec l’activité de la maladie dans d’autres conditions que celles pour lesquelles ils ont été conçus.
Les critères d’évaluation doivent aussi compter, c’est-à-dire qu’ils doivent refléter un réel changement dans le bien-être d’un patient. Par exemple, les essais sur la PR comprennent souvent des mesures de l’érosion et, quoique des différences minimales cliniquement importantes aient été établies, les répercussions cliniques de petites différences dans les scores d’érosion, même lorsqu’elles sont significativement différentes du comparateur d’un point de vue statistique, ne sont pas toujours évidentes30. Avant le début de l’étude, les questionnaires devraient si possible être validés dans le cadre de la maladie étudiée pour confirmer qu’un résultat positif est réellement en corrélation avec un changement dans l’état du patient.
Les critères d’évaluation devraient aussi refléter un niveau approprié de suivi pour la maladie traitée. Les résultats préliminaires peuvent sous-estimer ou surestimer les résultats d’un traitement à long terme. En particulier, les études utilisant le score mSASSS (Modified Stoke Ankylosing Spondylitis Spinal Score) pour mesurer les lésions visibles à la radiographie chez les patients souffrant de spondylarthrite ankylosante ont besoin d’un suivi à long terme pour voir des résultats utiles.
Finalement, les critères d’évaluation peuvent compter des événements indésirables et la façon dont ils sont déclarés est importante. La fréquence de tels événements peut être exagérée ou minimisée en étant exprimée de façon différente : en pourcentage de tous les patients, en chiffres absolus, en chiffres par 100 années-patient et ainsi de suite. Assurez-vous que la fréquence des événements comprenne ceux pour toute la durée de l’essai, puisque qu'ils n’apparaissent pas tous soudainement.

Les résultats
Le premier diagramme dans un rapport d’essai clinique est souvent un diagramme du roulement des patients, qui indique le nombre de patients recrutés, ceux exclus de la participation, ceux qui ont été répartis aléatoirement dans chaque groupe de traitement et ceux suivis à des moments précis (Figure 1). Ce diagramme utile est un moyen rapide d’examiner comment la population étudiée a changé avec le temps et de déterminer combien de patients parmi ceux inscrits sont effectivement enregistrés dans les résultats.
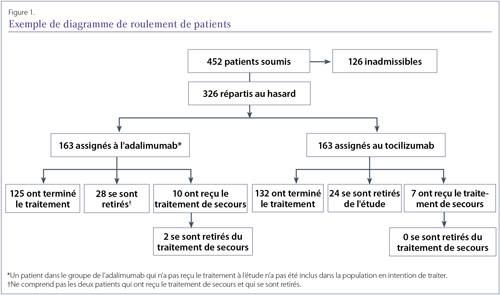
Un bon exemple d’un diagramme de roulement de patients de l’essai ADACTA, bien que des détails sur les raisons du retrait de patients l’amélioreraient25.
L’analyse des données
Une analyse par intention de traiter (ITT) examine les résultats pour tous les patients aléatoirement assignés à tout groupe de traitement, même s’ils ne reçoivent jamais de traitement. C’est la méthode d’analyse des données la moins biaisée31-33. Néanmoins, certains investigateurs effectuent plutôt une analyse en traitement reçu, qui compare les patients en fonction du traitement qu’ils ont reçu, ou une analyse per protocole, qui n’utilise que les données des sujets qui ont satisfait à tous les critères du protocole et qui ont terminé le traitement qui leur a été assigné. Non seulement la taille réduite des échantillons résultant de ces approches peut causer une perte de puissance statistique permettant de déceler les différences entre les traitements, elle amenuise aussi les bienfaits de la randomisation34.
Il existe plusieurs façons de gérer les données manquantes des patients qui se sont retirés de l’étude. Une approche rigoureuse est celle de l’imputation pour les non-répondants, qui présume que tout sujet pour qui il manque des données ne satisfait pas aux critères d’évaluation de l’étude. Une autre approche est l’imputation. Dans cette approche, les autres réponses des sujets sont utilisées pour estimer le ou les points de données manquants, même s’il est impossible de vérifier l’exactitude de ces estimations. Une troisième approche est l’analyse ayant recours à la dernière observation rapportée prospectivement (DORP), qui utilise les données les plus récentes du sujet au lieu de la donnée manquante. C’est une approche courante, mais puisque le manque de bénéfice clinique est la raison principale pour laquelle les patients se retirent, les analyses DORP ont tendance à exagérer les taux de réussite de tous les groupes de traitement.
Une approche encore plus rigoureuse que l’imputation pour les non-répondants a été utilisée par l’étude ORAL Standard concernant la comparaison du tofacitinib, de l’adalimumab et d’un placebo pour la PR35. Les patients étaient évalués pour non-réponse après trois mois, et les patients ne répondant pas au placebo étaient assignés à la thérapie active. Cependant, les patients non-répondants après trois mois dans un groupe de thérapie active ne pouvaient pas être caractérisés comme étant répondants à l’évaluation du critère principal après six mois, même s’ils étaient devenus répondants à ce moment-là. On appelle cela l’imputation des réponses nulles avec pénalité d’avancement. Le même plan a été utilisé dans l’essai FUTURE 2, qui portait sur l'utilisation du secukinumab dans l’arthrite psoriasique, et dans les essais MEASURE concernant le même médicament pour la spondylarthrite ankylosante, faisant paraître moins fiables les données avec ce plan d’essai sévère36,37.
Lorsqu’on examine des résultats, il est aussi utile de savoir quand les critères ont été mesurés. Certains essais peuvent se terminer de façon prématurée pour des raisons éthiques (p. ex. un traitement est apparu tellement meilleur que l’autre qu’il serait contraire à l’éthique de continuer à administrer aux patients le traitement inférieur), mais il est aussi possible de publier des résultats positifs concernant des points dans le temps, qui sont cependant provisoires et ne reflètent pas nécessairement les résultats finaux des essais. C’est ce qui s’est produit dans l’essai CLASS, qui portait sur le célécoxib par rapport aux AINS classiques, avec la publication de résultats positifs après 6 mois tandis que les résultats à 12 mois du critère d’évaluation de l’essai étaient négatifs38. Il faut prendre connaissance du protocole de l’essai pour voir si les résultats concernant tous les points dans le temps prévus à l’origine et critères d’évaluation indiqués dans le protocole sont présentés.
L’interprétation des résultats
La façon dont les données sont décrites influence la façon dont elles sont perçues. Par exemple, il a été démontré que les médecins sont plus susceptibles d’utiliser une thérapie si ses résultats d’essai sont présentés comme une réduction du risque relatif (le médicament A a réduit le risque de 40 % de plus que le médicament B) plutôt que comme une réduction absolue du risque (le médicament A a réduit le risque de 10 % à 5,8 % tandis que le médicament B a réduit le risque de 10 % à 7 %) ou un NST (le fait de traiter 83 patients avec le médicament A plutôt qu'avec le médicament B empêcherait la survenue d'un événement)39.
La présentation des données
Il existe plusieurs façons de montrer visuellement des données pour faciliter la lecture. Les courbes de survie de Kaplan-Meier sont utilisées fréquemment dans les dossiers d’essais cliniques en cardiologie et en oncologie, mais moins dans les articles sur la rhumatologie, qui ont tendance à utiliser des graphiques linéaires pour mettre en évidence les modifications à la mesure des résultats survenues au fil du temps. Cependant, les graphiques linéaires et les diagrammes à barres peuvent être manipulés, le plus souvent en n’incluant pas entièrement l’axe Y, ce qui peut faire en sorte que les différences de résultats peuvent sembler plus grandes qu’elles ne le sont en réalité.
Les graphiques en forêt (Figure 2) sont moins courants, mais ils sont particulièrement efficaces pour présenter les résultats entourant l’efficacité pour un éventail de critères, ou pour le même critère, dans plusieurs sous-groupes (ou plusieurs études dans le cas d’une méta-analyse). On utilise une ligne simple pour représenter chaque résultat, avec une boîte centrale qui représente l’estimation de l’effet moyen. Dans les graphiques de méta-analyse, la zone de la boîte peut varier afin de montrer le poids de chaque étude. La largeur de la ligne de chaque côté de la boîte affiche les intervalles de confiance pour ce résultat. Si elles croisent la ligne médiane verticale, qui peut représenter un risque relatif de 1 ou une différence de 0 entre les groupes, cela indique que le résultat n’est pas considéré comme statistiquement significatif puisque que le résultat réel pourrait se trouver d’un côté ou de l’autre de la ligne et, par conséquent, pourrait favoriser un côté ou l’autre.
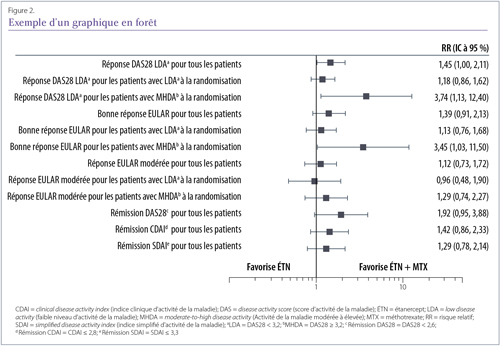
Exemple d’un graphique en forêt démontrant des résultats de risque relatif pour plusieurs critères dans l’étude Canadian Methotrexate and Etanercept Outcome Study. Lorsque la boîte noire se trouve du côté gauche de la ligne médiane, le résultat favorise l’étanercept; lorsqu’elle se trouve du côté droit, le résultat favorise l’étanercept plus le méthotrexate. La ligne médiane représente un risque relatif de 1, ce qui signifie qu’aucun effet n’a été observé.
Statistiques
D’un point de vue statistique, l'utilisation de la valeur p pour calculer si les résultats d'un groupe de traitement sont significativement différents de ceux d'un groupe de comparaison est une pratique courante depuis de nombreuses années. Toutefois, ces dernières années, les valeurs p ont fait l’objet d’un examen approfondi, car elles ne dépendent pas seulement des données, mais aussi des méthodes statistiques utilisées et des hypothèses avancées41. De plus, les valeurs p sont souvent interprétées à tort comme représentant la probabilité que l’hypothèse nulle s’avère vraie (c’est-à-dire qu'une valeur p de 0,04 signifie qu’il n’y a que 4 % de chances que l’hypothèse nulle soit vraie), plutôt que la probabilité qu’on obtienne ces résultats si l’hypothèse nulle était vraie (c’est-à-dire que s’il n’y avait aucune différence entre les groupes de traitement, une valeur p de 0,04 signifie qu'il n'y aurait que 4 % des chances d’obtenir ces résultats au hasard).
Un autre désavantage des valeurs p est que si suffisamment d’essais sont réalisés, certains seront positifs par hasard. Un plan d’essai avec un grand nombre de critères d’évaluation peut indiquer que les chercheurs espèrent qu’au moins un critère s’avérera statistiquement significatif selon la loi de la moyenne.
Certaines revues préfèrent maintenant9 que la signification statistique soit exprimée avec des intervalles de confiance (IC) qui indiquent une variation aléatoire autour d’une estimation ponctuelle. Contrairement aux valeurs p, le calcul des IC produit une valeur ponctuelle estimée et indique la plage de valeurs pour la population (pas seulement l’échantillon) qui pourrait produire cette valeur. Plutôt que de simplement rejeter ou soutenir une hypothèse nulle, les IC fournissent aussi de l’information sur la variabilité (précision) de la variable statistique et de son probable lien avec la population à partir de laquelle l’échantillon a été tiré42.
Les conclusions
Dans la section Discussion de certaines études, il n’est pas rare d'y lire des théories, émises par les auteurs d'études, sur les implications potentielles des résultats. Bien que de telles hypothèses puissent porter à réfléchir, elles peuvent diriger le lecteur vers des conclusions qui ne sont pas étayées par les données présentées dans le document. Par exemple, les auteurs d’un essai contrôlé par placebo peuvent discuter de leurs résultats par rapport à ceux d’un autre essai contrôlé par placebo même si les plans des deux essais sont peut-être très différents. Les essais comparatifs directs sont la seule manière de comparer deux interventions de façon fiable.
Les auteurs peuvent aussi discuter du fait que tous les autres essais publiés impliquant un agent spécifique ont démontré des effets précis. Ceci peut être une fausse hypothèse puisque les essais ayant des résultats positifs sont plus susceptibles d’être cités et publiés, en particulier dans des revues avec un plus grand facteur d’impact, que ceux avec des résultats négatifs. Les investigateurs peuvent se sentir obligés de publier seulement les documents ou les rapports dont les critères d’évaluation sont statistiquement significatifs7,41,43.
Des discussions à propos des effets de classe figurent souvent à la fin des documents puisqu’on a tendance à présumer que les médicaments avec le même mécanisme d’action, ou même ceux que l’on décrit comme faisant partie de la même catégorie, auront un effet semblable. Il est difficile de caractériser les effets de classe, et il n’existe aucune définition uniformément acceptée44-46. Par conséquent, il est difficile de déterminer si un effet de classe existe pour un ensemble de médicaments avant d’établir si cet effet est partagé par un agent en particulier. Quand il s’agit de prendre des décisions touchant les médicaments appartenant à une catégorie donnée (Tableau 2), il est suggéré que les cliniciens appliquent une hiérarchie des preuves plutôt que de présumer d’un effet de classe44,45. En général, il est judicieux de rechercher des preuves de l’efficacité et de l’innocuité d’un médicament en particulier dans des conditions spécifiées44-46, comme les organismes de réglementation le font.
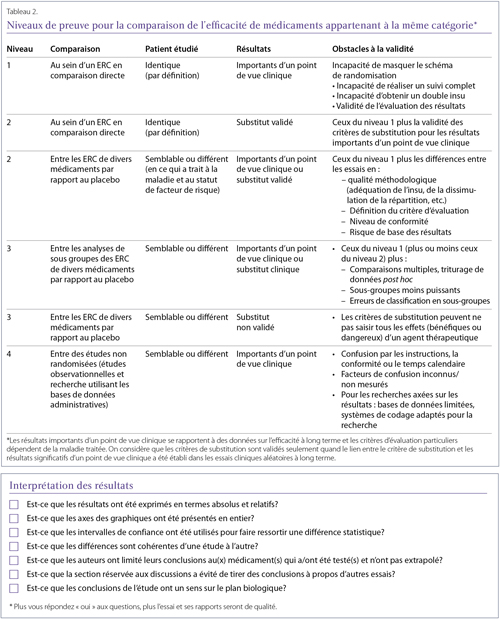
Discussion / résumé
Le présent article visait à analyser les différents facteurs qui déterminent si les résultats d’un essai clinique peuvent s’appliquer aux patients de la pratique d’un clinicien. Ceci n’est pas une analyse exhaustive, et plusieurs articles ont débattu des détails en profondeur. Cependant, nous espérons avoir fourni une introduction avec une liste de contrôle pour la qualité des essais cliniques aux rhumatologues praticiens leur permettant de mieux évaluer les rapports d’essai qui aboutissent sur leur bureau.
Philip Baer, M.D.C.M., FRCPC, FACR
Rhumatologue clinicien
Président, Section de la rhumatologie,
Association médicale de l'Ontario
Toronto (Ontario)
Michael Starr, M.D.
Professeur adjoint de médecine,
Université McGill
Rhumatologue,
Centre universitaire de santé McGill,
Montréal (Canada)
Nigil Haroon, M.D., Ph.D., D.M.
Clinicien scientifique,
Rhumatologue,
Réseau universitaire de santé
Toronto (Ontario)
Références :
1. Bykerk VP, et coll. Canadian Rheumatology Association recommendations for pharmacological management of rheumatoid arthritis with traditional and biologic disease-modifying antirheumatic drugs. J Rheumatol 2012;39:1559–82.
2. Papageorgiou SN, et al. Basic study design influences the results of orthodontic clinical investigations. J Clin Epidemiol 2015;68:1512-22.
3. Berbano EP, Baxi N. Impact of patient selection in various study designs: identifying potential bias in clinical results. South Med J 2012;105(3):149-55.
4. Dubost JJ, et coll. The changing face of septic arthritis complicating rheumatoid arthritis in the era of biotherapies. Retrospective single-center study over 35 years. Joint Bone Spine 2016 Jun 3. pii: S1297-319X(16)30061-6. doi: 10.1016/j.jbspin.2016.03.008. [Publication électronique avant impression]
5. Wood L, et coll. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336:601-5.
6. Colditz GA, et coll. How study design affects outcomes in comparisons of therapy. I: Medical. Stat Med 1989;8(4):441-54.
7. Stewart LA, Parmar MKB. Bias in the analysis and reporting of randomized controlled trials. Int J Technol Assess Health Care 1996;12(2):264-75.
8. Hollis S, et coll. Best practice for analysis of shared clinical trial data. BMC Med Res Methodol 2016;16 Suppl 1:76.
9. Ranstam J. Why the P-value culture is bad and confidence intervals a better alternative. Osteoarthritis Cartilage 2012;20:805-8.
10. Ludbrook J. Multiple comparison procedures updates. Clin Exp Pharmacol Physiol 1998;25:1032-7.
11. Vashisht P, Sayles H, Cannella AC, Mikuls TR, Michaud K. Generalizability of patients with rheumatoid arthritis in biologic clinical trials. Arthritis Care Res 2016;68:1478-88.
12. Bedaiwi MK, et coll. Clinical efficacy of celecoxib compared to acetaminophen in chronic nonspecific low back pain: results of a randomized controlled trial. Arthritis Care Res (Hoboken) 2016;68:845-52.
13. van der Heijde D, et coll. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: Two-year clinical and radiographic results from the TEMPO Study, a double-blind, randomized trial. Arthritis Rheum 2006;54(4):1063-74.
14. van der Heijde D, et coll. The safety and efficacy of adding etanercept to methotrexate or methotrexate to etanercept in moderately active rheumatoid arthritis patients previously treated with monotherapy. Ann Rheum Dis 2008;67:182-8.
15. Landewé R, et coll. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann Rheum Dis 2014;73:39-47.
16. Noordzij M, et coll. Sample size calculations. Nephron Clin Pract 2011;118:c319-23.
17. Malone HE, et coll. Fundamentals of estimating sample size. Nurse Res 2016;23(5):21-5.
18. Dijkman B, Kooistra B, Bhandari M. How to work with a subgroup analysis. Can J Surg 2009;52:515-22.
19. Keen HI, et coll. The prevalence of underpowered randomized clinical trials in rheumatology. J Rheumatol 2005;32(11):2083-8.
20. Soni A, et coll. Supervised exercise plus acupuncture for moderate to severe knee osteoarthritis: a small randomised controlled trial. Acupunct Med 2012;30(3):176-81.
21. Sieper J, et coll. Efficacy and safety of adalimumab in patients with non-radiographic axial spondyloarthritis: results of a randomised placebo-controlled trial (ABILITY-1). Ann Rheum Dis 2013;72(6):815-22.
22. Fleischmann R, et coll. Short-term efficacy of etanercept plus methotrexate vs combinations of disease-modifying anti-rheumatic drugs with methotrexate in established rheumatoid arthritis. Rheumatology (Oxford) 2014;53:1984-93.
23. Ministère de la Santé et des Services humains, Food and Drug Administration. Arthritis Advisory Committee Meeting. July 23, 2013. sBLA 125057/323: adalimumab for the treatment of Active non-radiographic axial spondyloarthritis in adults with objective signs of inflammation by elevated c-reactive protein (CRP) or magnetic resonance imaging (MRI), who have had an inadequate response to, or are intolerant to, a nonsteroidal anti-inflammatory drug. Disponible à l'adresse : http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM361563.pdf. Consulté le 5 mai 2016.
24. Estellat C, et coll. Control treatments in biologics trials of rheumatoid arthritis were often not deemed acceptable in the context of care. J Clin Epidemiol 2016;69:235-44.
25. Gabay C, et coll. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet 2013;381(9877):1541-50.
26. Willkens RF, et coll. Randomized, double-blinded, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. Arthritis Rheum 1984;27:376-86.
27. Nishimoto N, et coll. Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL-6 receptor inhibition therapy. Mod Rheumatol 2009; 19:12-19.
28. Narkus A, et coll. The placebo effect in allergen-specific immunotherapy trials. Clin Transl Allergy 2013;3(1):42.
29. Benedetti F, Dogue S. Different placebos, different mechanisms, different outcomes: lessons for clinical trials. PLoS One 2015;10(11):e0140967.
30. Bruynesteyn K, et coll. Determination of the minimal clinically important difference in rheumatoid arthritis joint damage of the Sharp/van der Heijde and Larsen/Scott scoring methods by clinical experts and comparison with the smallest detectable difference. Arthritis Rheum 2002;46:913-20.
31. Montori VM, Guyatt GH. Intention-to-treat principle. Can Med Assoc J 2001;165:1339-41.
32. Wang Y, et coll. Causal inference methods to assess safety upper bounds in randomized trials with noncompliance. Clin Trials 2015;12(3):265-75.
33. Baron G, et coll. Violation of the intent-to-treat principle and rate of missing data in superiority trials assessing structural outcomes in rheumatic diseases. Arthritis Rheum 2005;52(6):1858-65.
34. Lee YJ, et coll. Analysis of clinical trials by treatment actually received: is it really an option? Stat Med 1991;10(10):1595-605.
35. van Vollenhoven RF, et coll. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012; 367:508-19.
36. McInnes IB, et coll. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;386:1137-46.
37. Baeten D, et coll. Secukinumab, an interleukin-17a inhibitor, in ankylosing spondylitis. N Engl J Med 2015;373:2534-48.
38. Jüni P, Rutjes AW, Dieppe PA. Are selective COX2 inhibitors superior to traditional non steroidal anti-inflammatory drugs? BMJ 2002;324:1287-8.
39. Bobbio M, et coll. Completeness of reporting trial results: effect on physicians' willingness to prescribe. Lancet 1994;343(8907):1209-11.
40. Pope JE, et coll. The Canadian Methotrexate and Etanercept Outcome Study: a randomised trial of discontinuing versus continuing methotrexate after 6 months of etanercept and methotrexate therapy in rheumatoid arthritis. Ann Rheum Dis 2014;73:2144-51.
41. Chavalarias D, et coll. Evolution of reporting P values in the biomedical literature, 1990-2015. JAMA 2016;315(11):1141-8.
42. Sim J, Reid N. Statistical inference by confidence intervals: issues of interpretation and utilization. Phys Ther 1999;79(2):186-95.
43. Fanelli D. Do pressures to publish increase scientists' bias? An empirical support from US States Data. PLoS One 2010;5(4):e10271.
44. McAlister FA, Laupacis A, Wells GA, Sackett DL. User’s Guides to the Medical Literature: XIX. Applying clinical trial results B. Guidelines for determining whether a drug is exerting (more than) a class effect. JAMA 1999;282:1371-7.
45. Kennedy HL, Rosenson RS. Physicians’ interpretation of “class effects”: a need for thoughtful re-evaluation. J Am Coll Cardiol 2002;40:19-26.
46. Furberg CD. Class effects and evidence-based medicine. Clin Cardiol 2000;23(7 Suppl 4):IV15-9.
Glossaire
Ampleur de l’effet : La différence entre deux résultats divisée par l’écart-type de la population touchée. L’ampleur de l’effet se concentre sur l’importance de la différence de résultats plutôt que sur la taille des groupes de traitement.
Analyse en intention de traiter : Une analyse des résultats d’un essai qui comprend les données de chaque participant randomisé, même de ceux qui n’ont pas reçu le traitement.
Analyse intermédiaire : Une analyse planifiée qui compare les groupes d’un essai avant que celui-ci ne soit officiellement terminé. De cette façon, un essai peut être interrompu si la différence entre les groupes est si grande que les participants du groupe recevant l’intervention la moins efficace sont inutilement mis en danger.
Analyse ajustée : Une analyse qui rend compte des différences de base entre les caractéristiques importantes des patients (en les ajustant).
Attrition : La perte de participants en cours d’étude, aussi appelée perte de vue.
Censuré : Dans les études dans lesquelles le résultat correspond à un événement particulier, un terme décrivant le manque de données chez les patients dont le résultat est inconnu. Par exemple, si on sait qu’un patient est en vie seulement jusqu’à un certain point, le « temps de survie » est censuré à ce point.
Critère principal : La mesure de résultats considérée comme étant la plus importante pour évaluer l’effet d’une intervention.
Critère secondaire : Une mesure de résultats qui est moins importante que le critère principal, mais qui est toujours intéressante pour l’évaluer l’effet d’une intervention.
Critères de substitution : Marqueurs (souvent physiologiques ou biochimiques) qui peuvent être mesurés assez facilement et utilisés pour prédire ou représenter des résultats importants d’un point de vue clinique qui seraient autrement difficiles à mesurer.
Différence relative : La différence dans l’ampleur d’un résultat entre deux groupes, en tenant compte de leur taille. Elle est toujours exprimée sous forme de ratio ou pourcentage et non pas sous forme d’unités. Par exemple, si le médicament A réduit le résultat de 10 points et que le médicament B le réduit de 15 points, la différence relative est de 50 % (le médicament B réduit le résultat de 50 % de plus que le médicament A).
Double insu : Type d’insu dans lequel deux groupes, généralement des investigateurs et des patients, ignorent quels patients ont reçu quelle intervention.
Écart : La différence dans l’ampleur d’un résultat entre deux groupes. Par exemple, si le médicament A réduit le résultat de 10 points et le médicament B le réduit de 15 points, l’écart est de 5 points. Contraire : différence relative.
Écart-type : L’écart moyen entre un ensemble d’observations et leur valeur moyenne, qui indique l’étendue ou la dispersion des observations.
>Essai contrôlé : Un type d’essai clinique dans lequel les résultats sont comparés à un standard appelé « témoin ». Le témoin peut être une autre intervention (« traitement de référence »), un placebo (« groupe placebo ») ou des observations d’un essai antérieur (« groupe témoin historique »).
Essai d’équivalence : Un essai visant à déterminer si les effets de deux traitements ou plus diffèrent d’une quantité qui n’est pas cliniquement significative.
Essai de non-infériorité : Une version unilatérale d’un essai d’équivalence visant à déterminer si les effets d’un traitement ne sont pas pires que ceux d’un autre d’une quantité cliniquement significative.
Essai de supériorité : Un essai visant à déterminer si les effets d’une intervention sont plus importants que les effets d’une autre. Contraire : essai de non-infériorité.
Essai ouvert : Décrit un essai clinique dans lequel l’insu n’est pas utilisé. Toutes les parties concernées savent donc quelles interventions ont été assignées aux différents participants.
Étude cas-témoin : Une étude dans laquelle les patients atteints d’une maladie donnée sont « jumelés » à des cas témoins (population générale, patients avec une autre maladie, etc.). On compare ensuite les données entre les deux groupes, à la recherche de différences notables. Habituellement, elle est rétrospective et s’intéresse souvent aux causes de la maladie plutôt qu'au traitement.
Étude de cohorte : Une étude dans laquelle les groupes de personnes sont choisis en fonction de leur exposition à un agent spécifique ou de leur développement d’une maladie et leur santé à long terme est suivie. Elle peut être rétrospective.
Étude de phase I : Une étude habituellement effectuée avec des volontaires en bonne santé pour déterminer l’innocuité d’un médicament.
Étude de phase II : Une étude menée pour recueillir des données préliminaires sur l’efficacité d'un traitement chez les patients atteints d’une maladie précise.
Étude de phase III : Une étude menée pour recueillir plus d’information à propos de l’innocuité et l’efficacité d’un médicament en étudiant diverses populations, dosages et combinaisons de médicaments.
Étude de phase VI : Une étude qui survient après que les organismes de réglementation aient approuvé un médicament pour commercialisation. Elle est menée dans le but recueillir plus d’information sur le médicament.
Étude observationnelle : Une étude clinique dans laquelle les résultats des participants sont observés et évalués sans qu’ils soient assignés à des interventions spécifiques. Les études de cohorte et les études cas-témoin, entre autres, sont des études observationnelles.
Étude prospective : Une étude dans laquelle les participants sont désignés, puis suivis pendant un temps déterminé pour observer la survenue d'événements. Son contraire : l'étude rétrospective.
Étude rétrospective : Une étude dans laquelle les événements qui se sont produits chez les participants sont survenus avant qu’ils ne soient désignés comme faisant partie de l’essai.
Groupe expérimental : Le groupe de participants qui reçoit l’intervention visée par l’étude.
Groupe témoin : Un groupe de participants à l’étude qui ressemblent à ceux recevant l’intervention à l’essai, mais qui ne reçoivent pas cette intervention.
Hypothèse nulle : L’hypothèse qu’il n’y a pas de différence entre deux groupes. Les essais sont réalisés dans le but de réfuter l’hypothèse nulle et de démontrer qu’une différence réelle existe.
Important d’un point de vue clinique : Une description d’un effet suffisamment marqué pour revêtir une importance pratique pour les patients et pour les professionnels des soins de la santé.
Insu : Une procédure de plan d’essai dans laquelle un ou plusieurs groupes participant à l’essai (comme les patients, les investigateurs et les examinateurs externes) ignorent quels patients ont reçu quelle intervention.
Intervalle de confiance : Une mesure de l’incertitude entourant le résultat d’une analyse statistique. Un intervalle de confiance de 95 % (dont l’abréviation est IC à 95 %) signifie que si l’étude était faite à de nombreuses reprises avec d’autres groupes de la même population, 95 % des intervalles de confiance de ces études contiendraient la valeur réelle. Des intervalles de confiance élargis (p. ex. 90 %) indiquent moins de précision.
Intervention fictive : Une procédure ou un dispositif fait de telle façon qu’il est impossible de le distinguer de la procédure ou du dispositif à l’étude, mais qui ne contient pas de processus ou de composants actifs.
Limites de confiance : Les limites supérieures et inférieures d’un intervalle de confiance.
Nombre de sujets à traiter (NST) : Le nombre moyen de personnes qui doivent recevoir un traitement pour qu’une personne évite les résultats néfastes.
Nombre de sujets traités par sujet lésé : Le nombre moyen de personnes qui doivent être exposées à un facteur de risque pendant une période déterminée pour qu’une personne soit lésée par celui-ci.
Perte de vue : voir Attrition.
Plan d’étude croisé : Un plan d’essai dans lequel les groupes de participants reçoivent deux interventions ou plus dans un ordre précis. Par exemple, dans un plan croisé deux par deux, un groupe reçoit initialement le médicament A, puis le médicament B dans une phase ultérieure. L’autre groupe reçoit initialement le médicament B, puis le médicament A.
Plan factoriel : Un plan d’essai dans lequel de nombreux groupes de participants reçoivent une intervention parmi de nombreuses combinaisons. Par exemple, un plan factoriel deux par deux compte quatre groupes de participants. Chaque groupe pourrait recevoir une des combinaisons suivantes : médicament A et médicament B; médicament A et un placebo; médicament B et un placebo ou deux placebos. Dans cet exemple, toutes les combinaisons possibles des deux médicaments et du placebo sont étudiées dans un groupe de participants.
Plan parallèle : un plan d’essai dans lequel deux groupes de participants ou plus reçoivent différentes interventions au cours de la même période.
Point de départ : Le point initial dans une étude juste avant que les participants ne commencent à recevoir l’intervention que l’on teste.
Rapport de cotes (RC) : Le rapport de cotes d’un événement dans un groupe (habituellement le groupe de traitement) aux cotes de cet événement dans un autre groupe (habituellement le groupe témoin). Un rapport de cotes au-dessus de 1 signifie que le premier groupe est plus susceptible de vivre l’événement, tandis qu’un rapport de cotes en dessous de 1 suggère que cela est moins probable.
Rapport de risque : Un rapport comparant deux taux de risque (le temps qu’il faut avant qu’un événement ne se produise). Un rapport de risque au-dessus de 1 suggère que le groupe représenté par le premier numéro (habituellement le groupe de traitement) est plus susceptible de voir l’événement se produire sur une période de temps donnée que le second groupe (habituellement le groupe témoin). À la différence des rapports de cotes qui estiment la probabilité d’un événement cumulatif, les rapports de risque estiment la probabilité qu’un événement se produise à un moment précis.
Simple insu : Un type d’insu dans lequel un groupe de personnes participant à l’essai (patients, investigateurs ou examinateurs) ignorent quels patients ont reçu quelle intervention.
Statistiquement significatif : Improbable qu’il se soit produit uniquement par hasard. Mesuré par des tests statistiques qui calculent les valeurs p et les intervalles de confiance, entre autres résultats.
Valeur p : La probabilité (de 0 à 1) que le résultat observé ait pu être le fruit du hasard s’il n’y avait pas eu de différence entre les effets des interventions dans les groupes de l’essai.
|


