Printemps 2015 (volume 25, numéro 1)
Cette patiente est-elle atteinte d’une maladie associée aux IgG4?
par Luke Chen, M.D., FRCPC, MMEd
Télécharger le PDF
Cas : Une femme de 72 ans, originaire de l’Inde orientale, présente une protéinurie de catégorie néphrotique à 7,2 g/jour et un rapport albumine/créatinine (RAC) de 1,360 mg/mmol de créatinine. La biopsie rénale correspond à une glomérulonéphrite extra-membraneuse (GEM). Elle a un long historique de troubles cliniques rares commençant par une masse pancréatique à l’âge de 48 ans. Une histologie de la résection de Whipple a révélé un infiltrat lymphocytaire atypique sans indication de malignité; 15 ans plus tard, des biopsies de l’intestin grêle aux fins d’un bilan pour anémie révélaient un infiltrat lymphocytaire T atypique semblable. À 67 ans, elle présentait une pancréatite auto-immune répondant aux stéroïdes ainsi qu’une enflure bilatérale des glandes parotides et salivaires. Des biopsies excisionnelles de la glande salivaire ont révélé un « infiltrat lymphoplasmocytaire intense avec un nombre assez élevé de centres germinatifs (...) un fond modéré de fibrose et de sclérose. » La patiente a reçu un diagnostic de maladie de Mikulicz.
Son récent bilan sanguin indique une éosinophilie à 1,9 giga/L,
une concentration de protéines C-réactives (CRP) de 5,2 mg/L,
un résultat négatif au dosage des anticorps antinucléaires (ANA),
un faible taux d’albumine à 25 g/L, un taux élevé de protéines totales à 91 g/L avec un taux accru de globulines de 27 g/L (intervalle normal : 7 à 14 g/L) selon un modèle polyclonal avec un taux accru d’immunoglobuline G (IgG) à 41 g/L (intervalle normal : 6,3 à 18 g/L). Elle a été adressée pour déterminer s’il pouvait y avoir un trouble systémique expliquant la néphropathie actuelle et ses antécédents médicaux et si un tel diagnostic unificateur pourrait guider le choix d’un traitement systémique optimal pour sa GEM.
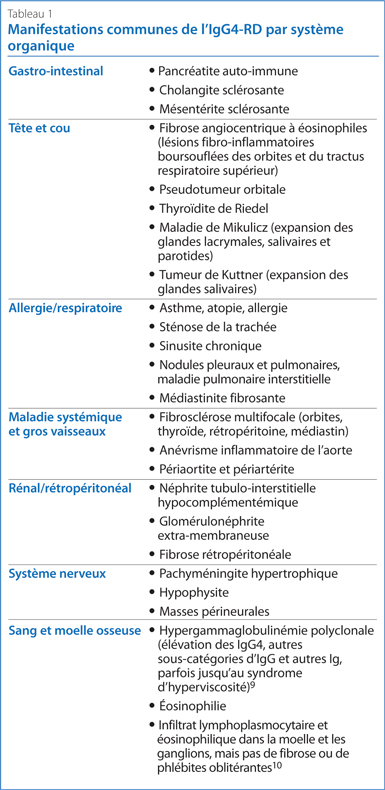
La maladie associée aux IgG4 (IgG4-RD) est une affection fibro-inflammatoire généralement associée à une tuméfaction (enflure) des tissus glandulaires, avec fibrose et infiltration des organes atteints par des lymphocytes polyclonaux, des cellules plasmatiques IgG4+ et des éosinophiles. Environ 70 % des patients présentent également des concentrations sériques d’IgG4 élevées. Pratiquement n'importe quel système organique peut être touché, mais comme pour les granulomes non caséifiés de la sarcoïdose, les résultats histologiques observés dans les différents tissus sont très similaires : fibrose « storiforme » (en forme de spirales irrégulières), phlébite oblitérante, infiltrat lymphoplasmocytaire et éosinophilie. L’IgG4-RD a été identifiée comme cause sous-jacente de nombreuses maladies avec lesquelles on ne voyait autrefois aucun lien, comme la pancréatite auto-immune1, la maladie de Mikulicz et la fibrosclérose multifocale. Le Tableau 1 énumère d’autres manifestations.
Cette brève revue se concentre sur trois questions :
• Quand devrais-je envisager l’IgG4-RD?
• Comment dois-je approcher le diagnostic de l’IgG4-RD?
• Comment dois-je traiter l’IgG4-RD?
La lecture de certaines revues plus approfondies est recommandée2-4.
Quand devrais-je envisager l’IgG4-RD?
Les patients présentent généralement une évolution subaiguë, souvent avec des manifestations pathologiques non reconnues. Les affections les plus fréquentes sont la pancréatite auto-immune, la sialoadénite, l’atteinte des glandes lacrymales et la fibrose rétropéritonéale. Le tiers des patients présenteront également de l’atopie, de l’asthme et de l’éosinophilie5.
Ce cas présente les manifestations classiques et la probabilité qu’il s’agisse d’IgG4-RD est extrêmement élevée. Ses caractéristiques d’éosinophilie, d’hypergammaglobulinémie polyclonale, d’IgG accrue, de CRP légèrement élevée et d’ANA négatif ou faiblement positif coïncident également avec l’IgG4-RD.
Comment dois-je approcher le diagnostic de l’IgG4-RD?
Lorsqu’on soupçonne la présence de l’IgG4-RD, les examens diagnostiques du Tableau 2 devraient être envisagés. Le plus important est l’examen pathologique des échantillons tissulaires. 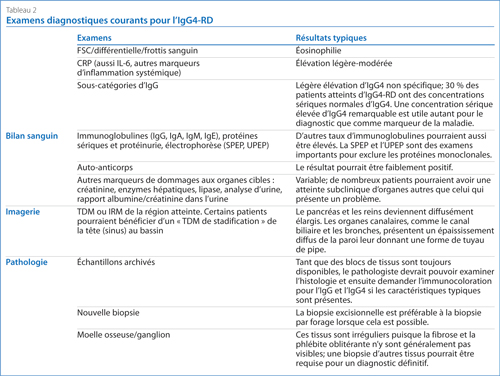
Cliquez ici pour obtenir une version agrandie de tableau
Dans de nombreux cas, des échantillons de tissu archivés seront disponibles; sinon, il conviendra de prélever un échantillon de l’organe le plus fortement atteint présentant le plus faible risque de morbidité liée à une biopsie. Une biopsie excisionnelle est préférable à une biopsie par forage. Une biopsie des glandes salivaires accessoires peut être envisagée chez les patients pour qui une biopsie des autres organes atteints (comme pour la fibrose rétropéritonéale) nécessiterait une laparotomie ou serait associée à un risque trop élevé. Si les résultats histologiques suggèrent une IgG4-RD, il conviendra de procéder à l'immunocoloration des IgG et des IgG4 (Figure 1).
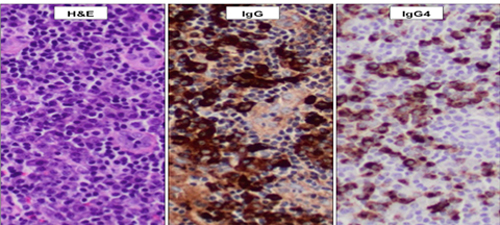
Figure 1.
Biopsie de ganglion, grossissement x 200.
Gauche : dense infiltrat de lymphocytes et de
cellules plasmatiques (« lymphoplasmocytaire »).
Centre : nombreuses cellules IgG4-positives.
Droite : nombreuses cellules IgG4-positives,
> 100/champ à fort grossissement et rapport IgG4/IgG > 40 %.
(Photo gracieusement offerte par le Dr Graham Slack, BC Cancer Agency.).
Les critères du consensus international6 s’appliquent, selon lesquels pour la plupart des tissus, une numération des IgG4 > 50 cfg-100/cfg et un rapport IgG4/IgG > 40 % accompagnés de résultats histologiques appropriés ont valeur de diagnostic. De nombreuses cellules IgG4-positives ne sont pas en soit spécifiques et peuvent être observées avec plusieurs « imitateurs » de l’IgG4-RD comme le syndrome de Sjögren, les lymphomes, d’autres tumeurs malignes, la vascularite et la maladie de Castleman. La pancréatite auto-immune classique avec observations radiologiques typiques est le seul cas où une confirmation histologique du diagnostic pourrait ne pas être nécessaire7.
La biopsie excisionnelle de la glande salivaire de la patiente effectuée en 1996 a été examinée et a confirmé « un infiltrat lymphoplasmocytaire marqué associé à une fibrose marquée. Le nombre de cellules
IgG4-positives est plus élevé et on observe de multiples champs à fort grossissement contenant plus de 100 cellules positives. Le rapport IgG4:IgG est supérieur à 40 %. » Le bilan sanguin a révélé une concentration sérique d’IgG4 élevée remarquable de 25,7 g/L avec d’autres sous-catégories normales ou légèrement élevées (normale
< 1,25 g/L). Les évaluations pathologiques et en laboratoire confirment le diagnostic d’IgG4-RD.
Comment dois-je traiter l’IgG4-RD?
Le traitement vise à atténuer les symptômes, à prévenir les dommages additionnels aux organes et à stabiliser la fibrose (qui est généralement irréversible). Le traitement initial se fait habituellement par la prednisone
1 mg/kg, laquelle est associée à un taux de réponse de 80 %8. La principale toxicité signalée est l’apparition ou l’aggravation du diabète puisque beaucoup de ces patients présentent une insuffisance pancréatique au départ. La déplétion des lymphocytes B avec le rituximab (1 gramme i.v. aux 2 semaines x 2 doses) est très efficace, réduisant les IgG4 par l’ablation des cellules plasmatiques à courte durée de vie qui les produisent11. La durée des périodes de rémission est variable et de nombreux patients devront être retraités. L’indice uniformisé des répondants IgG4-RD peut être utilisé pour le suivi des patients12. Les patients qui présentent une concentration sérique d’IgG4 élevée au départ disposent d’un moyen de suivi non invasif, bien que des rechutes puissent survenir même avec une concentration sérique d’IgG4 normale. La cytométrie de flux des plasmablastes en tant que marqueur plus sensible de l’activité de la maladie est à l’étude13.
Cette patiente a très bien répondu à la prednisone (1 mg/kg x
4 semaines, suivies d’une diminution progressive de la dose). Étant donné le fardeau de sa maladie et le risque de progression à une maladie rénale au stade terminal, elle a également reçu du rituximab (1 gramme i.v. x 3 doses). Elle demeure en rémission, avec des concentrations sériques normales d’éosinophiles, d’IgG, d’IgG4 et d’ACR avec le traitement d’entretien par la prednisone (5 mg/jour).
Références et lectures suggérées :
Pour plus d’information, veuillez écrire à lchen2@bccancer.bc.ca.
1. Kamisawa T, Funata N, Hayashi Y, et coll. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38(10):982-4.
2. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366(6):539-51.
3. Kamisawa T, Zen Y, Pillai S, et coll. IgG4-related disease. Lancet 2015 [publication électronique avant impression].
4. Mahajan VS, Mattoo H, Deshpande V, et coll. IgG4-related disease. Annu Rev Pathol 2014; 9:315-47.
5. Della Torre E, Mattoo H, Mahajan VS, et coll. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy 2014; 69(2):269-72.
6. Deshpande V, Zen Y, Chan JK, et coll. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012; 25(9):1181-92.
7. Shimosegawa T, Chari ST, Frulloni L, et coll. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas 2011; 40(3):352-8.
8. Masaki Y. Glucocorticoid Treatment in IgG4-RD. Présenté au International Symposium on IgG4-RD and Associated Conditions. Honolulu, Hawaii, 19 février 2014.
9. Chen LY, Wong PC, Noda S, et coll. Polyclonal hyperviscosity syndrome in IgG4-related disease and associated conditions. Clinical Case Reports 2015 Feb 2 [publication électronique avant impression].
10. Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol 2012; 29(4):226-34.
11. Carruthers MD, Topazian MD, Khosroshahi A, et coll. Rituximab for IgG4-Related Disease: A Prospective Clinical Trial. Arthritis Rheum 2013; 65(Suppl 10):2649.
12. Carruthers MN, Stone JH, Deshpande V, et coll. Development of an IgG4-RD Responder Index. Int J Rheumatol 2012; 2012:259408.
13. Wallace ZS, Mattoo H, Carruthers M, et coll. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis 2014; 74(1):190-5.
Luke Chen, M.D., FRCPC, MMEd
Professeur clinicien adjoint,
Division d’hématologie,
Université de la Colombie-Britannique
Hématologue,
Hôpital général de Vancouver
Vancouver, Colombie-Britannique |



